Перестановка Аза-Копа - Википедия - Aza-Cope rearrangement
Перестановки, особенно те, которые могут участвовать в каскадные реакции, Такие как перестановки аза-копа, имеют большое практическое, а также концептуальное значение в органическая химия, благодаря их способности быстро создавать сложные конструкции из простых исходных материалов. Аза-Коуп-перегруппировки являются примерами гетероатомных версий Справиться перестановкой, что является [3,3] -сигматропная перестройка что сдвигает одинарные и двойные связи между двумя аллильный составные части. В соответствии с правилами Вудворда-Хоффмана термические аза-коповские перегруппировки протекают супрафациально..[1] Перегруппировки Aza-Cope обычно классифицируются по положению азота в молекуле (см. Рисунок):

Первым примером аза-коповской перегруппировки была повсеместная катионная перегруппировка 2-аза-копе, который имеет место при температурах на 100-200 ° C ниже, чем перегруппировка Коупа из-за легкости перегруппировки.[2] Легкость этой перегруппировки объясняется как тем фактом, что катионный 2-аза-копе по своей природе является термонейтральным, что означает отсутствие предвзятости для исходного материала или продукта, так и наличием заряженный гетероатом в молекуле, что снижает активационный барьер. Менее распространены перегруппировка 1-аза-копе и перегруппировка 3-аза-копе, которые являются микроскопической обратной стороной друг друга. Перегруппировки 1- и 3-аза-копе имеют высокие барьеры активации и ограниченную синтетическую применимость, что объясняет их относительную неясность.[3][4][5]
Чтобы максимизировать его синтетическую полезность, катионная 2-аза-копе-перегруппировка обычно сочетается с термодинамическим смещением в одну сторону от перегруппировки. Наиболее распространенная и синтетически полезная стратегия объединяет катионная перегруппировка 2-аза-копе с циклизацией Манниха, и это тема большей части этой статьи. Эта тандемная реакция аза-Копа / Манниха характеризуется мягкими условиями реакции, диастереоселективностью и широкой синтетической применимостью. Обеспечивает легкий доступ к ацилзамещенным пирролидины, структура, обычно встречающаяся в натуральных продуктах, таких как алкалоиды, и был использован в синтезе ряда из них, особенно стрихнина и кринина.[6] Ларри Э. Оверман и коллеги провели обширное исследование этой реакции.[1]
Катионная перегруппировка 2-аза-копе

Катионная 2-аза-копе-перегруппировка, наиболее правильное название которой - 2-азония- [3,3] -сигматропная перегруппировка, была тщательно изучена Ларри Э. Оверманом и соавторами. Это наиболее широко изученная из перегруппировок аза-копе из-за мягких условий, требуемых для проведения компоновки, а также для ее многочисленных синтетических применений, особенно в синтезе алкалоидов. Термодинамически общая перегруппировка 2-аза-Копа не имеет смещения продукта, поскольку разорванные и образованные связи эквивалентны в любом направлении реакции, подобно перегруппировке Коупа. Наличие ионного гетероатома азота объясняет более легкая перегруппировка катионной перегруппировки 2-аза-копе по сравнению с перестановкой Коупа. Следовательно, он часто сочетается с термодинамическая мойка смещать продукт перегруппировки.[1]
В 1950 году Горовиц и Гейссман сообщили о первом примере перегруппировки 2-аза-копе, неожиданном результате неудачной попытки синтезировать аминоспирт.[2] Это открытие определило основной механизм перегруппировки, поскольку продукт, скорее всего, был получен посредством азотного аналога перегруппировки Коупа. Обработка аллилбензиламина (A) муравьиной кислотой и формальдегидом приводит к аминоспирту (B). Аминоспирт превращается в имин при добавлении кислоты (C), которая претерпевает катионную 2-аза-перегруппировку (D). Вода гидролизует иминиевый ион до амина (E). Обработка этого исходного материала только формальдегидом показала, что алкилирование аминогруппы происходит после катионной перегруппировки 2-аза-копе, что свидетельствует о быстрой легкости перегруппировки.[2]

Из-за мягких условий нагрева проводимой реакции, в отличие от более жестких условий для чисто углеводородной перегруппировки Копа, эта гетероатомная перегруппировка Коупа выдвинула гипотезу о том, что наличие положительного заряда на азоте в перегруппировке Копа значительно снижает активационный барьер для перестановка.[2]
Механизм реакции
Ускорение скорости за счет положительно заряженного азота

Перегруппировки аза-Коупа предсказываются Правила Вудворда-Хоффмана действовать надфасциально. Однако, хотя Оверман и его коллеги не исследовали это в явном виде, они выдвинули гипотезу, что, как и в случае катализируемого основанием окси-копе перегруппировка, заряженный атом искажает сигматропную перегруппировку с чисто согласованного механизма реакции (как и ожидалось в перегруппировке Коупа) к механизму с частичным бирадикальным / дипольным характером из-за делокализации положительного заряда на аллильном фрагменте, что ослабляет аллильную связь. Это приводит к снижению активационного барьера для разрыва связи. Таким образом, перегруппировка катион-аза-Коуп протекает быстрее, чем более согласованные процессы, такие как перегруппировка Коупа.[6][7]
Переходное состояние и стереохимия
Катионная перегруппировка 2-аза-копе характеризуется высокой стереоспецифичностью, которая возникает из-за ее высокого предпочтения переходному состоянию кресла. В своем исследовании стереоспецифичности этой перестройки Оверман и его коллеги использовали логику, аналогичную классическим экспериментам Деринга и Рота.[8] который показал, что перегруппировка Коупа предпочитает конформацию стула.[9] Используя катионную реакцию 2-аза-Копа / Манниха на предшественниках пирролизидина, они показали, что пирролизидины с цис-заместителями из E-алкенов и транс-заместителями из Z-алкенов в значительной степени предпочтительны, что свидетельствует о переходном состоянии стула. Если бы переходное состояние лодки действовало, были бы получены противоположные результаты (подробно показано на изображении ниже).[9] Как и во многих реакциях, превращение Z-енолята дает более низкую селективность из-за 1,3-диаксиальных стерических взаимодействий между енолятом и кольцом, а также того факта, что заместители предпочитают квазиэкваториальное расположение. Это помогает объяснить более высокие температуры, необходимые для превращения Z-енолята.[6][9]Перегруппировка катионо-2-аза-Коуп способствует переходному состоянию лодки даже в меньшей степени, чем перегруппировке Коупа: в аналогичных ситуациях, когда перегруппировка Коупа принимает переходное состояние лодочки, перегруппировка аза-Копа продолжается в кресле. геометрия.[1][6][10] Эти результаты согласуются с результатами вычислительной химии, которые дополнительно подтверждают, что переходное состояние находится под кинетическим контролем.[11]

Примечательно, что эти стереохимические эксперименты подразумевают, что катионная перегруппировка 2-аза-копе (а также циклизация Манниха) происходит быстрее, чем таутомеризация енола или иминия. Если бы это было не так, никакой значимой стереохимии не наблюдалось бы, что подчеркивает легкость этой быстрой реакции.[1]
Дополнительные соображения по стереохимии
Реакция аза-Копа / Манниха при участии в разжимные кольца, следует стереохимии, продиктованной наиболее благоприятной конформацией кресла, которая обычно размещает объемные заместители квазиэкваториально. При установке на кольцо виниловые и аминные компоненты могут иметь синхронные или антивирусные связи. Это соотношение обычно продиктовано аминовым заместителем: объемные заместители приводят к предшественникам синаза-копе.. Пока анти виниловые и аминные заместители обычно имеют только одно предпочтительное переходное состояние, приводящее к цис-конденсированной кольцевой системе, предпочтительный продукт син-заместителей может изменяться, что диктуется стерическими взаимодействиями с растворителями или большими N-заместителями, которые могут иметь преимущество перед объемными заместителями и изменять переходное состояние.[12][13]
Для простых реакций аза-Копа / Манниха, которые не участвуют в аннелировании с расширением кольца, а именно конденсации аминоспиртов и простых эфиров, вращение связи происходит быстрее, чем циклизация Манниха, и наблюдаются рацемические продукты.[14] Этого можно избежать, используя хиральный вспомогательный заместитель в амине. Реакции, связанные с кольцами, не могут подвергаться этим поворотам связей.[1]

Возможные термодинамические поглотители для смещения продукта перегруппировки
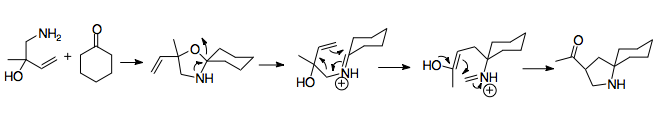
Первый пример Горовица и Гейссмана демонстрирует возможный термодинамический сток для связи с катионной перегруппировкой 2-аза-копе, где продукт смещается фенильным заместителем из-за арильной конъюгации, а затем захватывается гидролизом иминия. Другие методы смещения продукта включают использование заместителей, которые более стабильны по замещенным атомам углерода, высвобождение кольцевой деформации (например, путем объединения перегруппировки с раскрытием циклопропана), внутримолекулярный захват (на фото) и соединение перегруппировки с циклизацией Манниха.[1][15]

Реакция Аза-Копа / Манниха
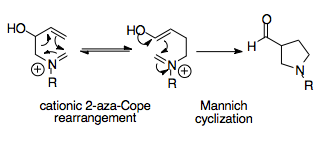
Реакция аза-Копа / Манниха является синтетически мощной реакцией, поскольку она способна создавать сложные циклические молекулы из простых исходных материалов. Эта тандемная реакция обеспечивает термодинамическое смещение в сторону одного продукта перегруппировки, поскольку циклизация Манниха необратима и ее продукт, ацилзамещенный пирролидин кольцо, более стабильное, чем у перестановки.[1][16]
Первая реакция аза-Копа / Манниха
Оверман и его коллеги признали, что катионная перегруппировка 2-аза-копе потенциально может быть синтетически мощной, если будет введен соответствующий термодинамический сток. Их логика заключалась в том, чтобы включить в исходный материал нуклеофильный заместитель, а именно спиртовую группу, которая действует только после перегруппировки, превращаясь в энол настроен для атаки иминиума.
Это первое сообщение о реакции было реакцией между альдегидами и 2-алкокси-3-бутенаминами, в результате которой образовывался аминоспирт, продукт реакции аза-Копа / Манниха которого представлял собой ацилзамещенное пирролидиновое кольцо. Эта простая процедура включала лишь умеренное нагревание в течение нескольких часов. Важно отметить, что реакция аза-Копа / Манниха протекает в одну стадию с отличным выходом. Эту процедуру легко применить для конденсации аминоэфиров (показано ниже), где сначала метилируют спирт.[16] После проведения реакции аза-Копа / Манниха кетон образуется путем добавления NaOH.[16] В этом простом случае амин не может образовывать иминиевый ион из основных кетонов; последующие методы нашли способы включения кетонов в реакцию.[16][17] Полезность этой реакции очевидна в том факте, что даже когда образуется менее стабильный изомер, реакция протекает, демонстрируя ее высокую термодинамическую благоприятность.[12][17]

Механизм реакции

Общий продукт реакции может потенциально происходить двумя отдельными путями: реакция аза-Копа / Манниха или циклизация аза-Принса /пинакольная перегруппировка. Эти механизмы обладают разными стереохимическими свойствами, которые объясняют преобладание реакции аза-Копа / Манниха. Реакция аза-Копа / Манниха заставляет каждый атом в аналоге [1,5] диена подвергаться sp2 гибридизация, стирающая стереохимию исходного материала в помеченном R 'положении, в то время как перегруппировка аза-принс / пинакол сохраняет стереохимию в помеченном R' положении, указывая на простой тест, который выявляет активный механизм. Энантиомерно чистый исходный материал в положении «R '» должен приводить к рацемическому продукту, если доминирующим механизмом является реакция аза-Копа / Манниха, в то время как стереохимия должна сохраняться, если доминирующим механизмом является циклизация аза-Принса / перегруппировка пинакола. путь. Простой эксперимент подтвердил, что продукт был рацемическим, что явилось очевидным доказательством того, что аза-коповская реакция Манниха является действующим механизмом. Дальнейшие эксперименты подтвердили это, используя знания о том, что ион карбения, образующийся в пути аза-Prins / пинакол, будет зависеть от способности его заместителя стабилизировать его положительный заряд, тем самым изменяя реактивность пути. Однако было показано, что различные заместители мало влияют на результат реакции, что снова указывает на аза-коповскую реакцию Манниха как на действующий механизм.[14]Недавняя литература из лаборатории Шанахана поддерживает редкий путь аза-Prins / пинакол, связанный только со значительно повышенной нуклеофильностью алкена и электрофильностью иминиума.[1][6][18][19]
Реакция аза-Копа / Манниха показывает высокую диастереоселективность, как правило, в соответствии с результатами стереохимические эксперименты, выясняющие переходное состояние катионной перегруппировки 2-аза-копе, что следует из того, что этот путь тандемной реакции был неотъемлемой частью этих экспериментов. Стереохимия перегруппировки несколько усложняется, когда заместители аллила и амина установлены на кольце и, таким образом, цис- или транс-друг к другу.
Исходный материал реакции аза-Копа / Манниха, аминоспирт, также можно рассматривать как связанный с окси-копе перегруппировка (ниже), как для ускорения его скорости из-за ионного участия, так и для аналогичной функции коллапса енола циклизации Манниха и таутомеризации кето-енола в перегруппировке окси-копе.[7]

Синтетические приложения реакции 2-аза-Копа / Манниха
Реакция аза-Копа / Манниха часто является наиболее эффективным способом синтеза пирролидин кольца, и, таким образом, имеет ряд применений в синтезе натуральных продуктов. Из-за своей диастереоселективности эта реакция была добавлена в каталог инструментов асимметричного синтеза, как видно на многих примерах асимметричного синтеза. алкалоиды синтезирован с использованием реакции. Как мы видели в первая реакция аза-Копа / Манниха и в выяснении реакции стереохимия, реакция аза-Копа / Манниха может быть использована для образования пирролидин кольца и пирролизидин кольца. Его можно использовать для создания многих дополнительных кольцевых структур, полезных в синтезе, таких как индолизидин циклы и индол кольца.[1][7]
(-) - Полный синтез стрихнина
Классическим примером, демонстрирующим полезность этой реакции, является синтез стрихнина Оверменом. Стрихнин это природный очень ядовитый алкалоид, встречается в роде древесных и вьющихся кустарников Стрихнос. Стрихнин обычно используется в качестве пестицида для мелких позвоночных. Первый полный синтез стрихнина, автор Р. Б. Вудворд,[20] представляет собой важный шаг в синтезе натуральных продуктов: ни одна молекула, приближающаяся к его сложности, не была синтезирована ранее. О следующем полном синтезе не сообщалось до конца 1980-х годов с использованием аналогичных методов, а именно с использованием промежуточного продукта, доступного в результате деградации стрихнина. Все эти синтезы использовались в суровых условиях. Синтез Овермана обходит эти проблемы и представляет собой первый асимметричный полный синтез стрихнина, использующий диастереоселективность и мягкие условия реакции аза-Копа / реакции Манниха. Стадия реакции аза-копа / Манниха протекала с почти количественным выходом. Соответственно, синтез Овермана на несколько порядков эффективнее своих предшественников.[20]

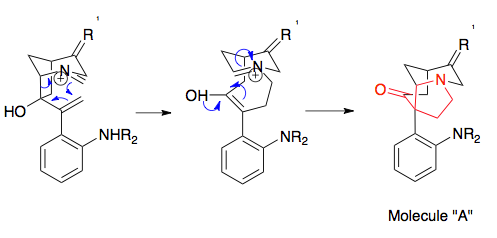
Синтез стрихнина Оверманом представляет собой полезный пример получения прекурсоров, необходимых для перегруппировки аза-Копа / Манниха, представляющий эффективное использование раскрытие эпоксидного кольца. Ключевые этапы синтеза субстрата перегруппировки, ведущего к исходным материалам, необходимым для реакции аза-Копа / Манниха, включали реакцию Стилле для соединения двух предшественников, эпоксидирование двойной связи с использованием трет-бутилгидропероксид, а Реакция Виттига для превращения кетона в алкен и стадию циклизации. Алкилирование амина (не показано) превращает молекулу в субстрат перегруппировки. Важно отметить, что эта молекула демонстрирует энантиомерную точность реакции аза-Копа / Манниха, поскольку простой энантиомерный исходный материал определяет конечный энантиомер: энантиомер стрихнина был получен с использованием энантиомера исходного материала.[20][21]

Синтез Овермана с подробным описанием синтеза субстрата перегруппировки, а также заключительных стадий реакции подробно описан здесь: Сверхчеловеческий синтез (-) - стрихнина.
Синтез (-) - кринина
Кринин это алкалоид семейства Amaryllidaceae, и его асимметричный полный синтез был одним из первых с использованием реакции аза-Копа / Манниха. Этот синтез представляет собой значительный шаг в развитии реакции аза-Копа / Манниха, так как он использует несколько наиболее полезных синтетических стратегий, характерных для данной реакции. В этой реакции используется высокая диастереоселективность катионной 2-аза-копе-перегруппировки, а также использование цианометильная группа для защиты амина во время виниллитий добавление и в качестве уходящей группы, чтобы способствовать образованию иминия, чему способствует добавление нитрат серебра.[22]Этот синтез является одним из примеров многих цианометильная группа обеспечение синтетически полезного пути образования пирролидина и индолизидина.

Синтез мостиковых трициклических алкалоидов
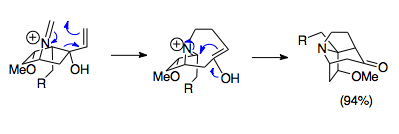
Оверман и его коллеги разработали методы синтеза сложных мостиковых трициклических структур с использованием реакции аза-Копа / Манниха. Эти аза-трициклические структуры находятся в комплексе Стемона семейство алкалоидов, а также в потенциальных лекарствах, таких как иммунодепрессанты. Показанный пример представляет собой легкую реакцию, объединяющую исходную соль 1-азабицикло [2.2.1] гептана с параформальдегидом при 80 ° C с образованием основной азатрициклической структуры Стемона молекулы алкалоидов. В частности, несмотря на неблагоприятное перекрытие орбиталей из-за стерических свойств этой кольцевой системы, реакция протекает с выходом 94%, что подчеркивает эффективность этой реакции даже в неблагоприятных условиях.[23]
Общее раскрытие и расширение кольца

Реакция аза-Копа / Манниха в сочетании с существующими кольцевыми циклами часто используется для создания индолизидин циклы (пирролидин, связанный с циклогексановым кольцом). Это типичное кольцо аннулирование, где циклопентановый фрагмент открывается при перегруппировке и закрывается при циклизации Манниха с образованием шестичленного кольца, присоединенного к пирролидиновому кольцу, в то время как наиболее популярное аннулирование аза-Копа / Манниха не единственное. Семичленные кольцевые циклы также можно синтезировать, так как ионы енола и иминия находятся в достаточно близкой близости, чтобы подвергаться циклизации Манниха.[22] О синтезе макроциклов с использованием этой реакции не сообщалось из-за отсутствия близости между енолом и иминием.[6]Винилоксазолидины также можно использовать в качестве субстратов для перегруппировки. Эта перестановка сначала создает винилоксазолидин от воздействия на циклогексанон аминобутенолом, который затем подвергается реакции аза-Копа / Манниха с использованием тепла и кислоты (по Льюису или протону). В этом примере разрывается, а затем образуется пятичленное кольцо. Более сложные примеры присоединяют оксазолидин к другому кольцу, представляя дополнительные способы образования индолизидиновых циклов.[24]
Сфера действия реакции аза-Копа / Манниха
Реакция аза-Копа / Манниха имеет множество преимуществ по сравнению с другими методами. Мягкие условия реакции не соблюдаются: легкий нагрев, обычно не выше 80 ° C, широкий диапазон растворителей и добавление 1 стехиометрического эквивалента кислоты, обычно Камфорсульфоновая кислота (CSA) или кислотой Льюиса. Другие пути к синтезу пирролидина не могут конкурировать со стереоспецифичностью, широкое применение в структурах, содержащих производные пирролидина, а также большой спектр возможных исходных материалов. Реакция проявляет высокая диастереоселективность, и является надежным, продолжая даже при плохом перекрытии орбит в переходном состоянии.[1]

Преимущества реакции аза-Копа / Манниха мотивировали исследования по синтезу исходных материалов для реакции, которые разделились на две основные категории: присоединение амина и образование иминия (красный) и установка винилового заместителя (синий). В реакции можно использовать широкий спектр N-заместителей (R), алкил и арил, некоторые из которых влияют на стереохимический результат реакции. Виниловые группы обычно ограничиваются 1,1- или 1,2-дизамещенными (винил с заместителями при R1, а R1,Р2 соответственно), с широким диапазоном допустимых электронных и стерических вариаций.[1]
Добавление амина и образование иминия
Открытие эпоксидного кольца
Кольцевая деформация эпоксидов обеспечивает полезную методологию для установки аминогруппы на два атома от спиртовой группы. Эпоксид может быть сначала разрушен нуклеофильной атакой бромида. Первичные амины, ароматические амины или анилиды лития также могут использоваться в качестве нуклеофилов. За этим этапом часто следует защитное О-метилирование, которое легко протекает.

Когда стерические свойства позволяют атаковать только соответствующий углерод (концевой углерод в отличие от второго углерода), прямое воздействие внутримолекулярного азота эффективно, как в случае с синтез стрихнина.[16][25]
Образование иминиум-иона
Наиболее распространенный способ генерировать иминиевый ион из установленного амина - это добавление формальдегид или же параформальдегид, который подвергается кислотно-каталитическому конденсация с образованием иминия. Синтез стрихнина Овермана олицетворяет этот метод.[6][25] Иногда используются внутримолекулярные карбонилы.[9] Другие методы образования иминиевых ионов включают использование цианометильные группы или используя оксазолидины как предшественники карбонила.
Алкилирование амина
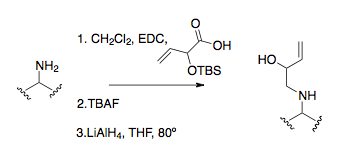
Алкилирование амина представляет собой распространенный метод получения предшественников имина. Алкилирование амина прямым SN2 реакция только изредка полезна для получения исходных материалов из-за высокой склонности аминов к сверхалкилату.[25]Восстановительное аминирование - более распространенная и эффективная процедура алкилирования, типичный в первой аза-коповской перегруппировке.[16][26][27]Самый полезный и стандартный метод алкилирования амина состоит в том, чтобы амин образовал амидную связь с последующим ее восстановлением, часто с LiAlH4.[9]
Использование оксазолидина
Кетоны и стерически затрудненные альдегиды не подходят для основной реакции аза-Копа / Манниха, поскольку амин не может образовывать с ними иминиевый ион. Дегидративное образование оксазолина с последующим нагреванием в присутствии полного эквивалента кислоты представляет способ обойти эту проблему. Оверман сообщил об использовании оксизолидинов для генерации иона иминия, необходимого для реакции. После образования Оверман показал, что циклогексаноны могут использоваться в качестве карбонильного компонента в синтезе пирролидина.[17] Эта реакция протекает с различными формами циклогексанонов. Когда был замещен ациклический кетон, реакция протекала с низким выходом, что подчеркивает термодинамическую благоприятность высвобождения циклогексанона из карбонила с двойной связью, поскольку это создает неблагоприятную деформацию связи в конформации кресла. Это одна из самых удобных конструкций кольцевой системы 1-азаспиро [4,5] декана, полезного природного продукта.[17]
Установка винилового заместителя
Винилирование кетонов
Винилирование может дать дополнительные синтетические преимущества, позволяющие расширить функциональность реакции.[23] Литийорганические реагенты обычно используются. Часто к азоту добавляют заместитель или защитную группу, хотя это не всегда необходимо. Добавление лития в реакцию оказывает большое влияние на стереохимию исходного материала, поскольку азот координируется с ним. Исходные материалы, на которые влияет эта координация, обычно приводят к предшественникам анти-аза-копе, тогда как те, которые не являются предшественниками, например, содержащие сильно замещенные стерически затрудненные амины, приводят к предшественникам син. Таким образом, природа заместителя азота имеет большое значение..[6][25]
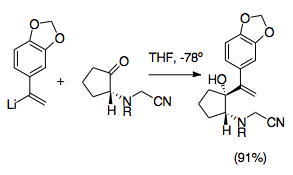
Использование цианометильной группы
Цианометильные группы представляют собой простой способ защитить иминиевый ион во время аллильного винилирования кетона. Цианамид группы и аналоги часто использовались при генерации ионов иминия. Обычно их устанавливают путем нуклеофильного присоединения к иону имина, обычно полученного алкилированием амина формальдегидом. Таким образом, иминиевый ион замаскирован.[28] Отсюда следует, что использование цианометильной группы обеспечивает эффективный способ контроля реакции аза-Копа / Манниха. Цианометильная группа защищает азот в положении 2 во время образования другого аллильного аналога по логике, аналогичной цианидному типу. умполунг. Затем он позже обеспечивает хорошую уходящую группу для образования иминиевого иона в соответствии с его использованием в генерации иминиевого иона.[29] Генерация иминиевых ионов из цианометильных групп обычно стимулируется добавлением нитрата серебра, хотя использовались и другие соединения серебра и меди. Этот дополнительный шаг позволяет более точно контролировать генерацию иминиевых ионов.[6][29] Важно отметить, что эти подготовительные реакции должны проводиться при -78 ° C, чтобы предотвратить взаимодействие цианометил / виниллитий. Этот метод также позволяет использовать множество различных возможных N-заместителей и может использоваться для упрощения синтеза октагидроиндолов и пирролы.[1][29]

1- и 3-аза-копе-перегруппировки

1- и 3-аза-копе-перегруппировки неясны по сравнению с катионными 2-аза-копе-перегруппировками из-за их энергий активации, которые сравнительно намного выше, чем у катионных 2-аза-копе-перегруппировок.
1- и 3-аза-копе имеют тенденцию к образованию имина в отличие от образования енамина, поскольку π-связь углерод-азот сильнее, чем σ-связь углерод-азот, что означает, что перегруппировка 3-аза-копе является термодинамически благоприятной, в то время как перегруппировка 1-аза-копе - нет: имин почти на 10 ккал / моль меньше по энергии. Таким образом, большие активационные барьеры 3-аза-коупа основаны на кинетике.[30] Исследования перегруппировок 1 и 3-аза-копе были сосредоточены на поиске хороших движущих сил для снижения барьеров активации. Несколько версий этих перегруппировок были оптимизированы для синтетической утилиты. Перегруппировка 1-аза-Коуп обычно сопряжена с термодинамическими движущими силами. Перегруппировки 3-аза-копе обычно проводят катионно, чтобы снизить кинетический барьер для его термодинамически благоприятного продукта.[30][31]
Эти перестановки следуют за большей частью Механистическая логика катионной перегруппировки 2-Aza-Cope. Перегруппировки 1- и 3-аза-копе происходят преимущественно через переходные состояния кресла (и сохраняют стереохимию, как и катионная перегруппировка 2-аза-копе) и являются ускорился с введением положительного заряда, так как это придает переходному состоянию более бирадикальный / диполярный характер.[31] Ожидается, что перегруппировка 3-аза-копе (и, следовательно, также перегруппировка 1-аза-копе, которая проходит через то же переходное состояние) будет проявлять еще менее ароматический характер в своем переходном состоянии по сравнению с перегруппировкой Коуп и катионным-2- aza-Cope перегруппировка, способствующая более высоким требуемым температурам (близким к температурам, необходимым для Cope перегруппировки, иногда даже выше, от 170 до 300 градусов) для преодоления кинетических барьеров активации для этих устройств.[3][31][32]
Перегруппировка 3-аза-копе

Реакция 3-аза-копе была обнаружена вскоре после того, как была идентифицирована перегруппировка 2-аза-копе, из-за ее аналогичной связи с перегруппировкой Клайзена. В самом деле, в ранних работах эту версию аза-коуповской перегруппировки часто называют перегруппировкой амино-Клайзена, что является искажением перегруппировки, поскольку это означает, что в молекуле присутствуют и азот, и кислород.[3] Эту перегруппировку можно использовать для образования гетероциклических колец с участием углерода, чаще всего пиперидина.
Один из первых примеров такого расположения был идентифицирован Бёрпиттом, который обнаружил перегруппировку, происходящую в солях аммония, которые из-за их заряженной природы протекали экзотермически без добавления тепла - что важно, без тетразамещенного азота перегруппировка не происходила.[33] Следуя этой логике, большая часть исследований перегруппировки 3-аза-Коупа была сосредоточена на заряженных цвиттерионных версиях этой реакции, поскольку распределение заряда помогает снизить барьер активации: в некоторых случаях перегруппировка может происходить при таких низких температурах, как - 20 ° С.[30][34]
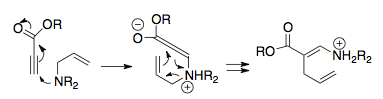
Хилл и Гилман впервые сообщили об общей незаряженной перегруппировке 3-аза-копе в 1967 году. После создания соответствующим образом замещенных енаминов интенсивное нагревание привело к почти полной перегруппировке иминного продукта. However, this rearrangement pathway has limited utility.[35]
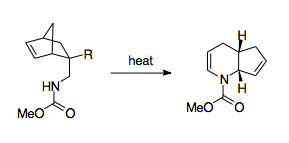
The 1-aza-Cope rearrangement

The first discovered 1-aza-Cope reaction was a simple analog to the generic Cope reaction and required intense heat to overcome its large thermodynamic activation barrier; most subsequent work on the 1-aza-Cope rearrangement has thus focused on pairing the arrangement with a driving thermodynamic force to avoid these harsh reaction conditions. It has been hypothesized that the 1-aza-Cope rearrangement rate-determining transition state has partial diradical and dipolar transition state character due to the presence of the heteroatom.[4]
Fowler and coworkers have come up with a scheme that mobilizes the 1-aza-Cope rearrangement as a synthetically useful route.[3] Fowler and coworkers recognized that because the barrier for the reaction lies in the nitrogen's thermodynamic preference to stay as an imine, stabilizing the nitrogen could have a thermodynamically beneficial effect. To that end, Fowler and coworkers installed a carbonyl group on the nitrogen, hypothesizing that the lone pair of the nitrogen would be stabilized by participation in an amide bond, and that the electronegativity of this amide group would lower the LUMO of the imine group, making the transition state more favorable.[3] Using this strategy, Fowler and coworkers were able to use the 1-aza-Cope rearrangement to create пиперидин и пиридин производные. This strategy was shown to be relatively robust, allowing for the formation of products even when forced through a boat transition state, when perturbed with substituent effects, or put in competition with alternative rearrangements.[3] Also significant is the relative ease of production of the reactants, which uses a Diels-Alder reaction paired with relatively simple workup steps, allowing for syntheses using complex cycling.[3]
Other methods of overcoming this thermodynamic barrier include pairing it with cyclopropane ring strain release, which allows the reaction to proceed at much lower temperatures.[30][36]

Смотрите также
- Reviews by Overman[1][37] и Siegfried Blechert.[38]
Рекомендации
- ^ а б c d е ж грамм час я j k л м п Overman, L. E.; Humphreys, P. G.; Welmaker, G. S. (2011). "The Aza-Cope/Mannich Reaction". Органические реакции. 75. pp. 747–820. Дои:10.1002/0471264180.or075.04. ISBN 978-0471264187.
- ^ а б c d Horowitz, R. M.; Geissman, T. A. (1950). "A Cleavage Reaction of α-Allylbenzylamines". Варенье. Chem. Soc. 72 (4): 1518–1522. Дои:10.1021/ja01160a025.
- ^ а б c d е ж грамм Chu M.; Wu P.L.; Givre S.; Fowler F.W. (1986). "The 1-AZA-Cope rearrangement". Буквы Тетраэдра. 27 (4): 461–464. Дои:10.1016/S0040-4039(00)85505-7.
- ^ а б Wu, P.L; Fowler, F. W. (1988). "The 1-aza-Cope rearrangement. 2". Журнал органической химии. 53 (26): 5998–6005. Дои:10.1021/jo00261a003.
- ^ Cook G.R.; Barta N.S.; Stille J.R. (1992). "Lewis acid-promoted 3-aza-Cope rearrangement of N-alkyl-N-allyl enamines". Журнал органической химии. 57 (2): 461–467. Дои:10.1021/jo00028a016.
- ^ а б c d е ж грамм час я Overman, L.E.; Mendelson, L. T.; Jacobsen, E. J. (1983). "Synthesis applications of aza-Cope rearrangements. 12. Applications of cationic aza-Cope rearrangements for alkaloid synthesis. Stereoselective preparation of cis-3a-aryloctahydroindoles and a new short route to Amaryllidaceae alkaloids". Варенье. Chem. Soc. 105 (22): 6629–6637. Дои:10.1021/ja00360a014.
- ^ а б c Overman, L. E. (1992). "Charge as a key component in reaction design. The invention of cationic cyclization reactions of importance in synthesis". Соотв. Chem. Res. 25 (8): 352–359. Дои:10.1021/ar00020a005.
- ^ Doering, W.v.E.; Roth, W. R. (1962). "The overlap of two allyl radicals or a four-centered transition state in the cope rearrangement". Тетраэдр. 18 (1): 67–74. Дои:10.1016/0040-4020(62)80025-8.
- ^ а б c d е Doedens, R. J.; Meier, G.P.; Оверман, Л. (1988). "Synthesis applications of cationic aza-Cope rearrangements. Part 17. Transition-state geometry of [3,3]-sigmatropic rearrangements of iminium ions". J. Org. Chem. 53 (3): 685–690. Дои:10.1021/jo00238a039.
- ^ Vogel, E.; Grimme, W.; Dinne, E. (December 1963). "Thermal Equilibrium between cis-1,2-Divinylcyclo-pentane and cis,cis-1,5-Cyclononadiene". Angewandte Chemie International Edition на английском языке. 2 (12): 739–740. Дои:10.1002/anie.196307392.
- ^ Lukowski M.; Jacobs K.; Hsueh P.; Lindsay H.A; Milletti M.C. (2009). "Thermodynamic and kinetic factors in the aza-Cope rearrangement of a series of iminium cations". Тетраэдр. 65 (50): 10311–10316. Дои:10.1016/j.tet.2009.10.010.
- ^ а б McCann, S. F.; Overman, L. E. (1987). "Medium Effects and the Nature of the Rate-Determining Step in Mannich-Type Cyclizations". Варенье. Chem. Soc. 109 (20): 6107–6114. Дои:10.1021/ja00254a033.
- ^ Overman, L. E.; Trenkle, W. C. (1997). "Controlling Stereoselection in Aza-Cope-Mannich Reactions". Isr. J. Chem. 37: 23–30. Дои:10.1002/ijch.199700005.
- ^ а б Jacobsen E. J.; Levin J.; Overman L. E. (1988). "Synthesis applications of cationic aza-Cope rearrangements. Part 18. Scope and mechanism of tandem cationic aza-Cope rearrangement-Mannich cyclization reactions". Варенье. Chem. Soc. 110 (13): 4329–4336. Дои:10.1021/ja00221a037.
- ^ Marshall, J. A.; Babler, J. H. (1969). "Heterolytic fragmentation of 1-substituted decahydroquinolines". J. Org. Chem. 34 (12): 4186–4188. Дои:10.1021/jo01264a104.
- ^ а б c d е ж Overman L. E.; Kakimoto, M. (1979). "Carbon-Carbon Bond Formation via Directed 2-Azonia-[3,3]-Sigmatropic Rearrangements. A New Pyrrolidine Synthesis". Варенье. Chem. Soc. 101 (5): 1310–1312. Дои:10.1021/ja00499a058.
- ^ а б c d Overman L.E.; Kakimoto M.; Okawara M. (1979). "Directed 2-azonia-[3,3]-sigmatropic rearrangements. a convenient preparation of substituted 1-azaspiro[4,5]decanes". Буквы Тетраэдра. 20 (42): 4041–4044. Дои:10.1016/s0040-4039(01)86498-4.
- ^ Armstrong, A.; Shanahan, S. E. (2005). "aza-Prins-pinacol approach to 7-azabicyclo[2.2.1]heptanes and ring expansion to [3.2.1]tropanes". Орг. Латыш. 7: 1335. Дои:10.1021/ja00221a037.
- ^ aza-Prins-pinacol approach to 7-azabicyclo[2.2.1]heptanes and ring expansion to [3.2.1]tropanes Armstrong, A.; Shanahan, S. E. Org. Lett. 2005, 7, 1335
- ^ а б c d R. B. Woodward; M. P. Cava; W. D. Ollis; A. Hunger; H. U. Daeniker; K. Schenker (1963). «Полный синтез стрихнина». Тетраэдр. 19 (2): 247–288. Дои:10.1016/S0040-4020(01)98529-1. PMID 13305562.
- ^ Knight, S.D.; Overman, L. E.; Pairaudeau, G. (1993). "Синтез катионных перегруппировок аза-копе. 26. Энантиоселективный полный синтез (-) - стрихнина". Варенье. Chem. Soc. 115 (20): 9293–9294. Дои:10.1021 / ja00073a057.
- ^ а б Overman, L. E.; Sugai, s. (1985). "Total Synthesis of (−)-Crinine. Use of Tandem Cationic Aza-Cope Rearrangement/Mannich Cyclizations for the Synthesis of Enantiomerically Pure Amaryllidaceae Alkaloids". Helv. Чим. Acta. 68 (3): 745–749. Дои:10.1002/hlca.19850680324.
- ^ а б Brueggemann, M.; McDonald, A. I.; Overman, L.E.; Rosen, M.D.; Schwink, L.; Scott, J.P. (2003). "Total Synthesis of (±)-Didehydrostemofoline (Asparagamine A) and (±)-Isodidehydrostemofoline". Варенье. Chem. Soc. 125 (50): 15284–15285. Дои:10.1021/ja0388820. PMID 14664560.
- ^ Overman, L. E.; Shim, J. (1993). "Synthesis applications of cationic aza-Cope rearrangements. Part 25. Total synthesis of Amaryllidaceae alkaloids of the 5,11-methanomorphanthridine type. Efficient total syntheses of (−)-pancracine and (.+-.)-pancracine". Органические реакции. 58 (17): 4662–4672. Дои:10.1021/jo00069a032.
- ^ а б c d Overman L. E.; Kakimoto, M.; Okazaki, M.E.; Meier, G.P. (1983). "Synthesis applications of aza-Cope rearrangements. 11. Carbon-carbon bond formation under mild conditions via tandem cationic aza-Cope rearrangement-Mannich reactions. A convenient synthesis of polysubstituted pyrrolidines". Варенье. Chem. Soc. 105 (22): 6622–6629. Дои:10.1021/ja00360a013.
- ^ Overman, L.E.; Fukaya, C. (1980). "Stereoselective total synthesis of (.+-.)-perhydrogephyrotoxin. Synthetic applications of directed 2-azonia-[3,3]-sigmatropic rearrangements". Варенье. Chem. Soc. 102 (4): 1454–1456. Дои:10.1021/ja00524a057.
- ^ Borch,R. F.; Bernstein, M. D.; Durst H. D. (1971). "Cyanohydridoborate anion as a selective reducing agent". Варенье. Chem. Soc. 93 (12): 2897–2904. Дои:10.1021/ja00741a013.
- ^ Grierson D. S.; Harris, M .; Husson, H.P. (1980). "Synthesis and chemistry of 5,6-dihydropyridinium salt adducts. Synthons for general electrophilic and nucleophilic substitution of the piperidine ring system". Варенье. Chem. Soc. 102 (3): 1064–1082. Дои:10.1021/ja00523a026.
- ^ а б c Overman, L. E.; Jacobsen, E. J. (1982). "The cyanomethyl group for nitrogen protection and iminium ion generation in ring-enlarging pyrrolidine annulations. A short synthesis of the amaryllidaceae alkaloid d,1-crinine". Tetrahedron Lett. 67 (51): 2741–2744. Дои:10.1016/S0040-4039(00)87446-8.
- ^ а б c d http://www.chem.uky.edu/research/cammers/thesis-pdf/2.pdf
- ^ а б c Jolidon, S.; Hansen, H. J. (1997). "Untersuchungen über aromatische Amino-Claisen-Umlagerungen". Helv. Чим. Acta. 60 (2): 978–1032. Дои:10.1002/hlca.19770600329.
- ^ Zahedi Ehsan; Ali-Asgari Safa; Keley Vahid (2010). "NBO and NICS analysis of the allylic rearrangements (the Cope and 3-aza-Cope rearrangements) of hexa-1,5-diene and N-vinylprop-2-en-1-amine: A DFT study". Central European Journal of Chemistry. 8 (5): 1097–1104. Дои:10.2478/s11532-010-0084-1.
- ^ Brannock Kent; Burpitt Robert (1961). "Notes- The Chemistry of Isobutenylamines. II. Alkylation with Allylic and Benzyl Halides". J. Org. Chem. 26 (9): 3576–3577. Дои:10.1021/jo01067a645.
- ^ Baxter, E. W.; Labaree, D.; Ammon, H. L.; Mariano, P. S. (1990). "Formal total synthesis of deserpidine demonstrating a versatile amino-Claisen rearrangement/Wenkert cyclization strategy for the preparation of functionalized yohimbane ring systems". Варенье. Chem. Soc. 12 (21): 7682–7692. Дои:10.1021/ja00177a032.
- ^ Hill, R. K.; Gilman, N. W. (1967). "A nitrogen analog of the Claisen rearrangement". Буквы Тетраэдра. 8 (15): 1421–1423. Дои:10.1016/S0040-4039(00)71596-6.
- ^ Boeckman, R. K.; Shair, M.D.; Vargas, R. J.; Stolz, L. A. (1993). "Synthetic and Mechanistic Studies of the retro-Claisen Rearrangement. 2. A Facile route to Medium-Ring Heterocycles via Rearrangement of Vinylcyclopropane- and Cyclobutanecarboxaldehydes". J. Org. Chem. 58 (2): 1295–1297. Дои:10.1021/jo00058a001.
- ^ Overman, L. E. (2009). "Molecular rearrangements in the construction of complex molecules". Тетраэдр. 65 (33): 6432–6446. Дои:10.1016/j.tet.2009.05.067. ЧВК 2902795. PMID 20640042.
- ^ Siegfried Blechert (1989). "The Hetero-Cope Rearrangement in Organic Synthesis". Синтез. 1989 (2): 71–82. Дои:10.1055/s-1989-27158.