Каскадная реакция - Cascade reaction

А каскадная реакция, также известный как реакция домино или же тандемная реакция, представляет собой химический процесс, который включает, по меньшей мере, две последовательные реакции, так что каждая последующая реакция происходит только благодаря химической функциональности, образованной на предыдущем этапе.[1] В каскадных реакциях выделение промежуточных продуктов не требуется, поскольку каждая реакция, составляющая последовательность, происходит спонтанно. В самом строгом определении этого термина условия реакции не меняются между последовательными стадиями каскада, и новые реагенты не добавляются после начальной стадии.[1][2] Напротив, одноразовые процедуры аналогичным образом позволяют проводить по меньшей мере две реакции последовательно без выделения промежуточных продуктов, но не препятствуют добавлению новых реагентов или изменению условий после первой реакции. Таким образом, любая каскадная реакция также является одноразовой процедурой, в то время как обратное неверно.[1] Хотя каскадные реакции часто состоят исключительно из внутримолекулярных превращений, они также могут происходить межмолекулярно, и в этом случае они также подпадают под категорию многокомпонентные реакции.[3]
Основные преимущества каскадных последовательностей включают высокую атомная экономика и сокращение количества отходов, образующихся в результате нескольких химических процессов, а также времени и работы, необходимых для их выполнения.[1][3][4] Эффективность и полезность каскадной реакции можно измерить с точки зрения количества связей, образующихся в общей последовательности, степени увеличения структурной сложности посредством процесса и ее применимости к более широким классам субстратов.[2][5]
Возможно, самый ранний пример каскадной реакции синтез тропинона, описанный в 1917 году Робинсоном.[6] С тех пор использование каскадных реакций в области полного синтеза расширилось. Точно так же значительно выросла разработка каскадной органической методологии. Этот повышенный интерес к каскадным последовательностям отражен в многочисленных соответствующих обзорных статьях, опубликованных за последние пару десятилетий.[1][2][3][4][5][7][8][9][10] Все большее внимание уделяется развитию асимметричного катализа каскадных процессов с использованием хиральных органокатализаторов или хиральных комплексов переходных металлов.[3][7][10][11]
Классификация каскадных реакций иногда бывает затруднительной из-за разнообразия этапов трансформации. К. С. Николау называет каскады нуклеофильными / электрофильными, радикальными, перициклическими или катализируемыми переходными металлами в зависимости от механизма участвующих стадий. В случаях, когда два или более класса реакции включаются в каскад, различие становится случайным, и процесс маркируется в соответствии с тем, что можно считать «основной темой».[4] Чтобы подчеркнуть замечательную синтетическую полезность каскадных реакций, большинство приведенных ниже примеров относятся к полному синтезу сложных молекул.
Нуклеофильные / электрофильные каскады
Нуклеофильные / электрофильные каскады определяются как каскадные последовательности, в которых ключевой этап представляет собой нуклеофильную или электрофильную атаку.[4]
Пример такого каскада виден в коротком энантиоселективном синтезе антибиотика широкого спектра действия (-) - хлорамфеникола, описанном Rao et al. (Схема 1).[3][12] Здесь хиральный эпоксидный спирт 1 сначала обрабатывали дихлорацетонитрилом в присутствии NaH. Получающееся промежуточное 2 затем прошел BF3· Et2О-опосредованная каскадная реакция. Внутримолекулярное раскрытие эпоксидного кольца дает промежуточное соединение 3, который после на месте гидролиз за счет избытка BF3· Et2O, предоставленный (-) - хлорамфеникол (4) с общей доходностью 71%.[3][12]

Нуклеофильный каскад также использовался в полном синтезе природного продукта пенталенена (схема 2).[4][13] В этой процедуре эфир скварата 5 обрабатывали (5-метилциклопент-1-ен-1-ил) литием и пропиниллитий. Две нуклеофильные атаки произошли преимущественно с транс в дополнение к промежуточным 6, который самопроизвольно претерпел 4π-вращательное электроциклическое раскрытие циклобутенового кольца. Полученные конъюгированные виды 7 уравновешен до конформера 8, который легче подвергался 8π-вращательной электроциклизации до сильно деформированного интермедиата 9. Потенциал высвобождения направленного штаммом протонирования 9 такой, что вид 10 был получен выборочно. Каскад завершился внутримолекулярной альдольной конденсацией, которая дала продукт 11 с общей доходностью 76%. Дальнейшая разработка дала целевой (±) -пенталенен (12).[4][13]

Органокаталитические каскады
Подкатегория нуклеофильных / электрофильных последовательностей состоит из органокаталитических каскадов, в которых основная нуклеофильная атака осуществляется за счет органокатализа.
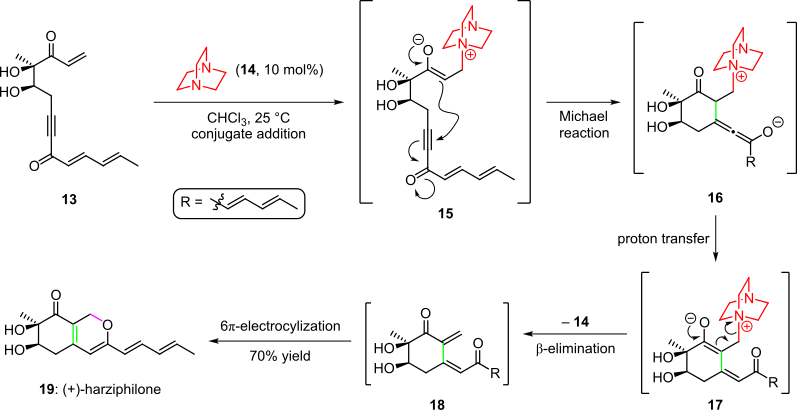
Органокаталитический каскад использовался в полном синтезе природного продукта гарцифилона, о чем сообщили Sorensen et al. в 2004 г. (схема 3).[4][14] Здесь обработка исходного материала енона 13 с органокатализатором 14 дал промежуточное звено 15 путем сопряженного добавления. Последующая циклизация путем внутримолекулярного присоединения по Михаэлю енолята к тройной связи системы дала частицы 16, что дало промежуточные 17 после переноса протона и таутомеризации. Каскад завершился удалением органокатализатора и спонтанным замыканием 6π-электроциклического кольца образовавшегося СНГ-диенон 18 к (+) - гарцифилон (19) с общей доходностью 70%.[4][14]
Выдающийся тройной органокаталитический каскад был описан Raabe et al. в 2006 г. Линейные альдегиды (20), нитроалкены (21) и α,β-ненасыщенные альдегиды (22) можно было бы сконденсировать вместе органокаталитически, чтобы получить тетра-замещенные циклогексанкарбальдегиды (24) с диастереоселективностью от умеренной до превосходной и полным энантиоконтролем (схема 4). Трансформация опосредуется легко доступным органокатализатором на основе пролина. 23.[15]

Превращение было предложено осуществлять посредством последовательности присоединения Майкла / добавления Майкла / альдольной конденсации (схема 5).[15] На первом этапе Майкл добавил альдегид 20 в нитроалкен 21 происходит посредством енаминового катализа с образованием нитроалкана 25. Конденсация α,β-ненасыщенный альдегид 22 с органокатализатором, затем облегчает конъюгированное добавление 25дать промежуточный енамин 26, который склонен к внутримолекулярной альдольной конденсации с образованием иминиума 27. Органокатализатор 23 регенерируется путем гидролиза вместе с продуктом 24, замыкая тройной каскадный цикл.[15]

Радикальные каскады
Радикальные каскады - это те, в которых ключевой этап представляет собой радикальную реакцию. Высокая реакционная способность свободных радикалов делает синтетические подходы на основе радикалов явно подходящими для каскадных реакций.[4]
Одним из наиболее широко известных примеров синтетической полезности радикальных каскадов является последовательность циклизации, использованная в полном синтезе (±) -гирсутена в 1985 году (схема 6).[4][16] Здесь алкилйодид 28 был преобразован в первичный радикал-промежуточный продукт 29, который прошел 5-экзо-запуск циклизации для получения реактивных частиц 30. Последующие 5-экзорадикальная циклизация приводит к промежуточной 31, который при закалке дал целевой (±) -гирсутен (32) с общей доходностью 80%.[4][16]

Каскадный радикальный процесс был также реализован в одном из полных синтезов (-) - морфина (схема 7).[4][17][18] Арилбромид 33 был преобразован в соответствующие радикальные виды 34 путем лечения три-п-бутилоловогидрид. А 5-экзо- затем произошла триггерная циклизация для получения промежуточных 35стереоселективно в силу стереохимии эфирной связи. На следующем этапе каскада геометрические ограничения 35 запретить кинетически благоприятные 5-экзо-триггерный путь циклизации; вместо вторичных бензильных радикалов 36 был получен с помощью агеометрически разрешенного 6-эндо-тригциклизация. Последующее отщепление фенилсульфинильного радикала дает продукт 37 с общим выходом 30%, который был далее переработан в (-) - морфин (38).[4][17][18]

Перициклические каскады
Перициклические реакции, возможно, наиболее часто встречающиеся в каскадных превращениях, включают циклоприсоединения, электроциклические реакции и сигматропные перегруппировки.[4] Хотя некоторые из вышеупомянутых примеров нуклеофильных / электрофильных и радикальных каскадов связаны с перициклическими процессами, этот раздел содержит только каскадные последовательности, которые состоят исключительно из перициклических реакций или в которых такая реакция, возможно, является ключевым этапом.
Типичным примером перициклического каскада является каскад эндиандриновой кислоты, описанный Nicolaou et al. в 1982 г. (схема 8).[4][19] Здесь высоконенасыщенная система 39 был впервые гидрирован до сопряженных тетраеновых форм 40, которые при нагревании претерпевают 8π-вращательное замыкание электроциклического цикла с образованием циклических промежуточных 41. Вторая спонтанная электроциклизация, на этот раз a6π-дисротационное замыкание кольца, преобразовала 41к бициклическим видам 42, геометрия и стереохимия которых благоприятствовали последующей внутримолекулярной реакции Дильса-Альдера. Метиловый эфир эндиандриновой кислоты B (43) таким образом получали с выходом 23%.[4][19]

Перициклическая последовательность, включающая реакции внутримолекулярного гетеро-циклоприсоединения, была использована в полном синтезе встречающегося в природе алкалоида (-) - виндорозина (схема 9).[4][20] Быстрый доступ к цели обеспечен раствором 1,3,4-оксадиазола. 44 в триизопропилбензоле, подвергающемся воздействию высоких температур и пониженного давления. Сначала произошла реакция гетеро-Дильса-Альдера с обратной потребностью электронов с образованием промежуточного соединения. 45.Термодинамически благоприятная потеря азота приводит к образованию 1,3-дипольных соединений. 46. Затем спонтанное внутримолекулярное [3 + 2] циклоприсоединение 1,3-диполя и индолесистемы формирует эндо-товар 47 с общей доходностью 78%. Дальнейшая работа дала целевой натуральный продукт. 48.[4][20]

Общий синтез (-) - коломбиазина А, о котором сообщила в 2005 г. группа Харровена, включал электроциклический каскад (схема 10).[4][21] Под воздействием тепла посредством микроволнового излучения производное скварата 49подверглись электроциклическому раскрытию циклобутенового кольца с последующим замыканием 6π-электроциклического кольца, в результате чего образовался бициклический промежуточный продукт. 51. Их таутомеризация дала ароматические частицы 52, который под воздействием воздуха окислился до продукта 53с общей доходностью 80%. Мишень (-) - коломбиазин А (54) был затем получен из 53с помощью реакции Дильса-Альдера при нагревании с последующим расщеплением терт-бутилзащитная группа.[4][21]

Некоторые [2,2] парациклофаны также могут быть получены виперициклическими каскадами, как сообщила группа Hopf в 1981 г. (Схема 11).[1][22] В этой последовательности реакция Дильса-Альдера между 1,2,4,5-гексатетраеном 55и диенофил 56 впервые образовал высокореактивный промежуточный продукт 57, который впоследствии димеризуется с образованием [2,2] парациклофана 58.[1][22]
![Схема 11. Перициклическая последовательность для синтеза [2,2] парациклофанов.](http://upload.wikimedia.org/wikipedia/commons/thumb/b/be/Scheme_11_-_peri_-_pcyclophane.svg/681px-Scheme_11_-_peri_-_pcyclophane.svg.png)
Каскады, катализируемые переходными металлами
Каскадные последовательности, катализируемые переходными металлами, сочетают новизну и мощность металлоорганической химии с синтетической полезностью и экономичностью каскадных реакций, обеспечивая еще более экологически и экономически желательный подход к органическому синтезу.[4]
Например, родиевый катализ был использован для превращения ациклических монотерпенов типа 59 до 4ЧАС-хроменовые продукты в каскаде гидроформилирования (схема 12).[8][23] Во-первых, селективное катализируемое родием гидроформилирование менее стерически затрудненной олефиновой связи в 59 давал ненасыщенный альдегид 60, который при тех же условиях превращался в промежуточный 61через карбониленовую реакцию. Второе катализируемое родием гидроформилирование до видов 62 с последующей конденсацией с образованием 4ЧАС-хромопродукты типа 63 с общей доходностью 40%.[8][23]

Родиевый катализ также использовался для инициирования каскада циклизации / циклоприсоединения в синтезе тиглиана, о котором сообщила группа Даубена (схема 13).[2][24] Лечение диазоимида 64 с димером ацетата родия (II) образовался карбеноид, который дал реактивный илид 65после внутримолекулярной циклизации с соседней карбонильной группой. Затем спонтанно возникло межмолекулярное [3 + 2] циклоприсоединение с образованием целевого тиглиана. 66.[2][24]

Формальное внутримолекулярное [4 + 2] циклоприсоединение 1,6-енинов типа 67опосредованный золотым катализом - еще один пример каскада, катализируемого переходными металлами (схема 14).[25][26] Различные 1,6-енины реагировали в мягких условиях в присутствии комплексов Au (I). 68а–б для получения трициклических продуктов 69 с урожайностью от умеренной до отличной.[25][26]
![Схема 14. Катализируемое золотом формальное внутримолекулярное [4 + 2] циклоприсоединение 1,6-енинов.](http://upload.wikimedia.org/wikipedia/commons/thumb/2/2b/Scheme_14_-_metal_-_gold_enyne.svg/481px-Scheme_14_-_metal_-_gold_enyne.svg.png)
Это формальное циклоприсоединение было предложено протекать через каскадный процесс, показанный на схеме 15.[25][26] Комплексообразование 1,6-енина67 с катионной формой катализатора дает промежуточные 70, в котором активированная тройная связь подвергается атаке олефиновой функциональности или замещенного циклопропана. 71. Электрофильное раскрытие трехчленного кольца образует катионные частицы. 72, который подвергается реакции типа Фриделя-Крафтса, а затем реароматизируется с образованием трициклического продукта 69.[25][26] Из-за природы взаимодействия комплексов золота с ненасыщенными системами этот процесс также можно рассматривать как электрофильный каскад.
![Схема 15. Предлагаемый каскадный процесс формального внутримолекулярного [4 + 2] циклоприсоединения 1,6-енинов.](http://upload.wikimedia.org/wikipedia/commons/thumb/b/ba/Scheme_15_-_metal_-_gold_enyne_mech.svg/751px-Scheme_15_-_metal_-_gold_enyne_mech.svg.png)
Примером каскадов, катализируемых палладием, является асимметричная полиеновая циклизация Хека, используемая при получении (+) - ксестохинона из трифлатного субстрата. 75 (Схема 16).[4][27] Окислительное присоединение арил-трифлатной связи к комплексу палладия (0) в присутствии хирального дифосфинового лиганда (S) -бинап дает хиральный комплекс палладия (II) 77. За этой стадией следует диссоциация трифлат-аниона, ассоциация соседнего олефина и 1,2-вставка нафтильной группы в олефин с образованием промежуточного соединения. 79. Вторая мигрирующая вставка в оставшуюся группу олефинов, за которой следует β- устранение, затем происходит получение продукта 81 с общим выходом 82% и с умеренной энантиоселективностью. На этой стадии также регенерируется палладиевый (0) катализатор, что позволяет повторно запустить каскад.[4][27]

Многоступенчатые тандемные реакции
Многоступенчатые тандемные реакции (или каскадные реакции) представляют собой последовательность химических превращений (обычно более двух этапов), которые происходят последовательно для превращения исходного материала в сложный продукт.[28] Подобные органические реакции предназначены для создания сложных структур, встречающихся в натуральный продукт полный синтез.
В общем синтезе спирокетала ионофор антибиотик routiennocin 1 (рис. 1), центральный спирокетальный скелет был построен с помощью многоступенчатой тандемной реакции (рис. 2).[29] Фрагмент A и фрагмент B были соединены в одну стадию с образованием ключевого промежуточного продукта G, который может быть дополнительно переработан с получением конечного продукта рутиенноцина.

В этой тандемной реакции произошло четыре химических превращения. Во-первых, обрабатывая фрагмент A н-бутиллитий образовал анион углерода, который атаковал алкилиодидную часть фрагмента B с образованием промежуточного соединения C (стадия 1). Затем производное 3,4-дигидропирана D образовывалось посредством опосредованного основанием реакция элиминации на промежуточном С (шаг 2). Защитная группа на 1, 3-диол фрагмент в промежуточном продукте D удаляли кислотной обработкой с получением диольного продукта E (стадия 3). Спирокетальный продукт G был получен внутримолекулярным путем. кеталь реакция образования. Эта многоступенчатая тандемная реакция значительно упростила построение этой сложной спирокетальной структуры и облегчила путь к полному синтезу рутиенноцина.

Рекомендации
- ^ а б c d е ж грамм час Титце, Л. Ф .; Бейфус, У. Энгью. Chem. Int. Эд. 1993, 32, 131–163.
- ^ а б c d е ж Padwa, A .; Бур, С.К. Тетраэдр 2007, 63, 5341–5378.
- ^ а б c d е ж грамм Пеллиссье, Х. Тетраэдр 2006, 62, 1619–1665.
- ^ а б c d е ж грамм час я j k л м п о п q р s т ты v ш Икс у z аа ab ac объявление ае Nicolaou, K. C .; Эдмондс, Д. Дж .; Балджер, П. Г. Энгью. Chem. Int. Эд. 2006, 45, 7134–7186.
- ^ а б Титце, Л.Ф. Chem. Ред. 1996, 96, 115–136.
- ^ Робинсон, Р. J. Chem. Soc. Пер. 1917, 111, 762.
- ^ а б Пеллиссье, Х. Тетраэдр 2006, 62, 2143–2173.
- ^ а б c d Wasilke, J.C .; Обри, С. Дж .; Baker, R.T .; Базан, Г.С. Chem. Ред. 2005, 105, 1001–1020.
- ^ Chapman, C .; Фрост, К. Синтез (Штутг). 2007, 2007, 1–21.
- ^ а б Эндерс, Д .; Grondal, C .; Хюттль, М. Р. М. Энгью. Chem. Int. Эд. 2007, 46, 1570–1581.
- ^ Grondal, C .; Jeanty, M .; Эндерс, Д. Nat. Chem. 2010, 2, 167–178.
- ^ а б Bhaskar, G .; Сатиш Кумар, В.; Венкатешвара Рао, Б. Тетраэдр: асимметрия 2004, 15, 1279–1283.
- ^ а б Paquette, L.A .; Гэн, Ф. Орг. Lett. 2002, 4, 4547–4549.
- ^ а б Stark, L.M .; Пекари, К .; Соренсен, Э. Дж. Proc. Natl. Акад. Sci. СОЕДИНЕННЫЕ ШТАТЫ АМЕРИКИ. 2004, 101, 12064–12066.
- ^ а б c d е Эндерс, Д .; Hüttl, M. R. M.; Grondal, C .; Раабе, Г. Природа 2006, 441, 861–863.
- ^ а б Curran, D. P .; Чен, М.-Х. Тетраэдр Латыш. 1985, 26, 4991–4994.
- ^ а б Паркер, К. А .; Фокас, Д. Дж. Являюсь. Chem. Soc. 1992, 114, 9688–9689.
- ^ а б Паркер, К. А .; Фокас, Д. Дж. Орг. Chem. 2006, 71, 449–455.
- ^ а б Nicolaou, K. C .; Petasis, N.A .; Зипкин, Р. Э .; Уениши, Дж. Варенье. Chem. Soc. 1982, 104,5555–5557.
- ^ а б Elliott, G.I .; Velcicky, J.; Ishikawa, H .; Li, Y .; Богер, Д. Л. Энгью. Chem. Int. Эд. 2006,45, 620–622.
- ^ а б Harrowven, D.C .; Pascoe, D.D .; Демуртас, Д .; Борн, Х. Энгью. Chem.Int. Эд. 2005, 44,1221–1222.
- ^ а б Hopf, H .; Бом, И.; Кляйншрот, Дж. Орг. Synth. 1981, 60, 41.
- ^ а б Roggenbuck, R .; Эйлбрахт, П. Тетраэдр Латыш. 1999, 40, 7455–7456.
- ^ а б Dauben, W. G .; Дингс, Дж.; Смит, Т.С. J. Org. Chem. 1993, 58, 7635–7637.
- ^ а б c d е ж Jiménez-Núñez, E .; Эчаваррен, А. М. Chem. Ред. 2008, 108, 3326.
- ^ а б c d Nieto-Oberhuber, C .; Лопес, С.; Эчаваррен, А.М. Варенье. Chem. Soc. 2005, 127, 6178–6179.
- ^ а б Maddaford, S.P .; Андерсен, Н.Г .; Cristofoli, W.A .; Кей, Б.А. Варенье. Chem. Soc. 1996, 118,10766–10773.
- ^ Nicolaou, K. C .; Эдмондс, Дэвид Дж .; Балджер, Пол Г. Энджью. Chem. Int. Эд. 2006, 45, 7134-7186.
- ^ Diez-Martin, D. Kotecha, N.R .; Ley, S. V .; Mantegani, S .; Menendez, J.C .; Орган, Х. М .; Уайт, А. Д., Tetrahedron, 1992, 48, 1899-7938.
внешняя ссылка
- Химические узлы в Периодическая таблица видео (Ноттингемский университет)