Дигидроксилирование - Dihydroxylation
Дигидроксилирование это процесс, посредством которого алкен превращается в вицинальный диол. Хотя для этого есть много способов окисление, наиболее распространенные и прямые процессы используют высокую степень окисления переходный металл (обычно осмий или марганец). Металл часто используют как катализатор, с некоторыми другими стехиометрический присутствует окислитель.[1] Кроме того, для катализа реакции были разработаны и использовались методы с использованием других переходных металлов и непереходных металлов.

Механизм

В механизме дигидроксилирования a лиганд сначала координируется с металлическим катализатором (обозначенным как осмий), который определяет хиральную селективность олефина. Затем алкен координируется с металлом посредством [3 + 2] циклоприсоединения, и лиганд диссоциирует от металлического катализатора. Гидролиз олефина затем образует вицинальный диол, а окисление катализатора стехиометрическим окислителем регенерирует металлический катализатор для повторения цикла.[2] Концентрация олефина имеет решающее значение для энантиомерного избытка диола, поскольку более высокие концентрации алкена могут связываться с другим каталитическим центром с образованием другого энантиомера.[3]
Катализируемые осмием реакции
Четырехокись осмия (OsO4) является популярным окислителем, используемым при дигидроксилировании алкенов из-за его надежности и эффективности при получении син-диолов. Поскольку он дорог и токсичен, каталитическое количество OsO4 используются вместе со стехиометрическим окислителем.[2][3] В Гидроксилирование Миласа, Дигидроксилирование Апджона, и Асимметричное дигидроксилирование по Шарплесу Во всех реакциях в качестве катализатора используется осмий, а также различные вторичные окислители.
Милас
Дигидроксилирование Миласа было введено в 1930 году и использует перекись водорода в качестве стехиометрического окислителя.[4] Хотя с помощью этого метода можно получить диолы, чрезмерное окисление дикарбонильного соединения привело к трудностям выделения вицинального диола.[4] Поэтому протокол Миласа был заменен асимметричным дигидроксилированием Апджона и Шарплесса.
Upjohn
О дигидроксилировании Апджона сообщалось в 1973 году, и в нем используется OsO4 в качестве активного катализатора в процедуре дигидроксилирования. Здесь также работают N-метилморфолин N-оксид (NMO) в качестве стехиометрического окислителя для регенерации осмиевого катализатора, что позволяет использовать каталитические количества осмия.[2][5] Протокол Upjohn обеспечивает высокую конверсию вицинального диола и допускает использование многих субстратов. Однако протокол не может дигидроксилировать тетразамещенные алкены.[2] Условия Апджона можно использовать для синтеза антидиолов из аллиловых спиртов, как продемонстрировали Киши с соавторами.[6]
Асимметричный шарплесс
Асимметричное дигидроксилирование по Шарплесу было разработано К. Барри Шарплессом для использования каталитических количеств OsO4 вместе со стехиометрическим окислителем K3[Fe (CN)6].[1][2][7] Реакцию проводят в присутствии хирального вспомогательного вещества. Выбор дигидрохинидина (DHQD) или дигидрохинина (DHQ) в качестве хирального вспомогательного вещества диктует лицевую селективность олефина, поскольку абсолютная конфигурация лигандов противоположна.[2][7][8] Катализатор, окислитель и хиральное вспомогательное вещество можно приобрести предварительно смешанными для селективного дигидроксилирования. AD-mix-α содержит хиральный вспомогательный компонент (DHQ)2PHAL, позиционирующий OsO4 на альфа-поверхности олефина; AD-mix-β содержит (DHQD)2PHAL и доставляет гидроксильные группы к бета-поверхности.[1][9] Асимметричное дигидроксилирование по Шарплесу имеет широкие возможности для селективности субстрата за счет изменения хирального вспомогательного класса.[7]


Другие варианты
Как упоминалось выше, способность синтезировать антидиолы из аллиловых спиртов может быть достигнута с использованием NMO в качестве стехиометрического окислителя.[6] Использование тетраметилендиамин (TMEDA) в качестве лиганда продуцировал синдиолы с благоприятным диастереомерным соотношением по сравнению с протоколом Киши; однако используется стехиометрический осмий. Син-селективность обусловлена донорной способностью аллилового спирта водородной связи и акцепторной способностью диамина.[10][11][12] С тех пор это применялось к гомоаллильным системам. [13]
Другие методы дигидроксилирования
Поскольку тетроксид осмия дорог и токсичен, для получения вицинальных диолов из олефинов использовали другие металлы. Еще один популярный металл, используемый при дигидроксилировании, - рутений. Несмотря на то, что он обладает высокой окислительной способностью, рутений использовался из-за его короткого времени реакции и экономической эффективности.[14] Обычно четырехокись рутения создается in situ из трихлорида рутения, а вторичный окислитель NaIO4 используется для регенерации катализатора. Стадия реакции, ограничивающая оборот, представляет собой стадию гидролиза; поэтому серная кислота добавляется для увеличения скорости этой стадии.[14][15]
Марганец также используется при дигидроксилировании и часто выбирается, когда методы четырехокиси осмия дают плохие результаты.[15] Как и у рутения, окислительный потенциал марганца высок, что приводит к чрезмерному окислению субстратов. Перманганат калия часто используется в качестве окислителя при дигидроксилировании; однако из-за его плохой растворимости в органическом растворителе катализатор межфазного переноса (например, хлорид бензилтриэтиламмония, TEBACl) также добавляется для увеличения количества субстратов для дигидроксилирования.[15]
Дигидроксилирование Превоста и Вудворда

В отличие от других описанных методов, в которых в качестве катализатора используются переходные металлы, в методах Превоста и Вудворда используются йод и соль серебра. Однако добавление воды в реакцию направляет цис- и транс-присоединение гидроксильных групп. В реакции Прево обычно используется бензоат серебра для получения транс-диолов; В модификации Вудворда реакции Прево для получения цис-диолов используется ацетат серебра. В обеих реакциях Превоста и Вудворда к алкену сначала добавляют йод, образуя циклический ион йода. Затем путем нуклеофильного замещения к иону йодиния добавляют анион соответствующей соли серебра.[16]

В реакции Прево ион йодиния подвергается нуклеофильной атаке бензоат-анионом. Бензоат-анион снова действует как нуклеофил, замещая йодид посредством механизма участия соседней группы. Второй бензоат-анион реагирует с промежуточным продуктом с образованием антизамещенного дибензоатного продукта, который затем может подвергаться гидролизу с образованием транс-диолов.[16]

Модификация Вудворда реакции Прево дает цис-диолы. Анион ацетата реагирует с циклическим ионом йодиния с образованием промежуточного иона оксония. Затем он может легко реагировать с водой с образованием моноацетата, который затем может быть гидролизован с образованием цис-диола. [17]
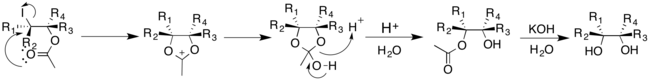
Чтобы исключить необходимость в солях серебра, Судалай и его сотрудники модифицировали реакцию Превоста-Вудворда; реакция катализируется LiBr и использует NaIO4 и PhI (OAc)2 как окислители.[18]LiBr реагирует с NaIO4 и уксусную кислоту для получения ацетата лития, который затем может протекать по реакции, как упоминалось ранее. Протокол показал высокое значение DR для соответствующего диола в зависимости от выбранного окислителя.

Приложения
Синтез высокозамещенных и стереоспецифических сахаров важен, поскольку полисахариды составляют большой класс соединений, встречающихся в природе. Одним из конкретных примеров является биологически активная молекула какелокелоза, которая, как было показано, обладает активностью против ВИЧ.[19] Исследования, проведенные Harris et al. работали над энантиоспецифическим синтезом сахаров, относящихся к какелокелозе и другим сахарам, с использованием множества различных реакций дигидроксилирования с осмиевым катализатором. Винилфуран подвергали реакции в условиях Шарплесса с AD-mix-α с получением (R) -диола. Позже в результате дигидропиран реагировал в условиях Апджона с получением полученного сахара, манноза (где R представляет собой либо H, либо защитную группу).[19]

Кроме того, таран и Gulose были также синтезированы из другого дигидропирана. Поскольку соединение содержит аллиловый спирт, условия Апджона и модификация Апджона с использованием TMEDA в качестве вторичного окислителя для создания образующихся сахаров (где R представляет собой H или защитную группу).[19]

Еще одно применение методов дигидроксилирования - синтез стероидов. Брассиностероиды - это класс стероидов, регулирующих рост растений, и было показано, что они обладают сельскохозяйственной активностью в качестве инсектицида. Этот класс стероидов содержит стандартную структуру стероидов в дополнение к четырем вицинальным диолам, которые имеют свою собственную стереохимию.[20] Броза установил гидроксильные группы в стероиде, используя оба условия Вудворда, чтобы получить цис-диол в кольцо А стероида. Затем алкеновую цепь на кольце D дигидроксилировали с получением второго цис-диола с использованием OsO4 и NMO в качестве стехиометрического окислителя.[21]

Рекомендации
- ^ а б c Кэри, Фрэнсис А.; Сандберг, Ричард Дж. Расширенная органическая химия, часть B: реакции и синтез (5-е изд.). Springer.
- ^ а б c d е ж Шредер, М. (1980). «Цис-гидроксилирование тетраоксида осмия ненасыщенных субстратов». Chem. Rev. 80 (2): 187–213. Дои:10.1021 / cr60324a003.
- ^ а б Kolbe, H.C .; VanNieuwanhze, M.S .; Шарплесс, К.Б. (1994). «Каталитическое асимметричное дигидроксилирование». Chem. Rev. 94 (8): 2483–2547. Дои:10.1021 / cr00032a009.
- ^ а б Milas, N.A .; Суссман, С. (1936). «Гидроксилирование двойной связи 1». Варенье. Chem. Soc. 58 (7): 1302–4. Дои:10.1021 / ja01298a065.
- ^ Dupau, P .; Epple, R .; Thomas, A.A .; Фокин, В.В .; Шарплесс, К.Б. (2002). "Осми-катализируемое дигидроксилирование олефинов в кислой среде: старый процесс, новые уловки". Adv. Synth. Катал. 344 (3–4): 421–33. Дои:10.1002 / 1615-4169 (200206) 344: 3/4 <421 :: AID-ADSC421> 3.0.CO; 2-F.
- ^ а б Cha, J.K .; Христос, W.J .; Киши, Ю. (1983). «1983». Tetrahedron Lett. 24: 3943–6. Дои:10.1016 / s0040-4039 (00) 88231-3.
- ^ а б c Morikawa, K .; Park, J .; Андерсон, П.Г .; Hashiyama, T .; Шарплесс, К.Б. (1993). «Каталитическое асимметричное дигидроксилирование тетразамещенных олефинов». Варенье. Chem. Soc. 115 (18): 8463–4. Дои:10.1021 / ja00071a072.
- ^ Carey, F.A .; Сундберг, Р.Дж. (2007). Расширенная органическая химия, часть A: структура и механизмы. Springer. п. 202.
- ^ Xu, D.X .; Crispino, G.A .; Шарплесс, К.Б. (1992). «Селективное асимметричное дигидроксилирование (АД) диенов». Варенье. Chem. Soc. 114 (19): 7570–1. Дои:10.1021 / ja00045a043.
- ^ Donohoe, T.J .; Лезвия, К .; Мур, П.Р .; Waring, M.J .; Winter, J.J.G .; Helliwell, M .; Ньюкомб, штат Нью-Джерси; Стемп, Г. (2002). «Направленное дигидроксилирование циклических аллиловых спиртов и трихлорацетамидов с использованием OsO4 / TMEDA». J. Org. Chem. 67 (23): 7946–56. Дои:10.1021 / jo026161y. PMID 12423122.
- ^ Donohoe, T.J .; Мур, П.Р .; Waring, M.J .; Ньюкомб, Николас Дж. (1997). «Направленное дигидроксилирование аллиловых спиртов». Tetrahedron Lett. 38 (28): 5027–30. Дои:10.1016 / s0040-4039 (97) 01061-7.
- ^ Донохо, Т.Дж. (2002). «Развитие направленной реакции дигидроксилирования». Synlett (8): 1223–32. Дои:10.1055 / с-2002-32947.
- ^ Донохо, Тимоти Дж .; Митчелл, Ли; Уоринг, Майкл Дж .; Хелливелл, Мадлен; Белл, Эндрю; Ньюкомб, Николас Дж. (10.06.2003). «Область направленного дигидроксилирования: приложение к циклическим гомоаллиловым спиртам и тригалогенацетамидам». Органическая и биомолекулярная химия. 1 (12): 2173–2186. Дои:10.1039 / B303081D. ISSN 1477-0539. PMID 12945911.
- ^ а б Plietker, B .; Ниггеманн, М. (2003). «Улучшенный протокол дигидроксилирования олефинов, катализируемого RuO4». Орг. Латыш. 5 (18): 3353–6. Дои:10.1021 / ol035335a. PMID 12943425.
- ^ а б c Bataille, C.J.R .; Донохо, Т.Дж. (2011). «Безосмиевое прямое син-дигидроксилирование алкенов». Chem. Soc. Rev. 40 (1): 114–28. Дои:10.1039 / b923880h. PMID 21049111.
- ^ а б Kurti, L .; Чако, Б. (2005). Стратегические применения названных реакций в органическом синтезе. Эльзевир. С. 360–1.
- ^ Woodward, R.B .; Brutcher, Jr., F.V. (1958). «Цис-гидроксилирование синтетического стероидного промежуточного соединения с йодом, ацетатом серебра и влажной уксусной кислотой». Варенье. Chem. Soc. 80: 209–11. Дои:10.1021 / ja01534a053.
- ^ Emmanuvel, L .; Shaikh, T.M.A .; Судалай, А. (2005). «NaIO4 / Li Br-опосредованное диастереоселективное дигидроксилирование олефинов: каталитический подход к реакции Превоста-Вудворда ». Орг. Латыш. 7 (22): 5071–4. Дои:10.1021 / ol052080n. PMID 16235960.
- ^ а б c Harris, J.M .; Керанен, доктор медицины; О'Догерти, Г.А. (1999). "Синтезы D- и L-Манноза, гулоза и талоза через реакции диастереоселективного и энантиоселективного дигидроксилирования ». J. Org. Chem. 64 (9): 2982–3. Дои:10.1021 / jo990410. PMID 11674384.
- ^ Bishop, G .; Конц, Чаба (2002). «Сигнализация брассиностероидов и стероидных гормонов растений». Растительная клетка. 14: S97–110. Дои:10.1105 / tpc.001461. ЧВК 151250. PMID 12045272.
- ^ Brosa, C .; Нусимович, С; Перакаула, Р. (1994). «Синтез новых брассиностероидов с потенциальной активностью как антиэкдистероиды». Стероиды. 59 (8): 463–7. Дои:10.1016 / 0039-128x (94) 90058-2. PMID 7985206. S2CID 45409677.