Синдром Герстмана – Штройсслера – Шейнкера - Gerstmann–Sträussler–Scheinker syndrome
| Синдром Герстмана – Штройсслера – Шейнкера | |
|---|---|
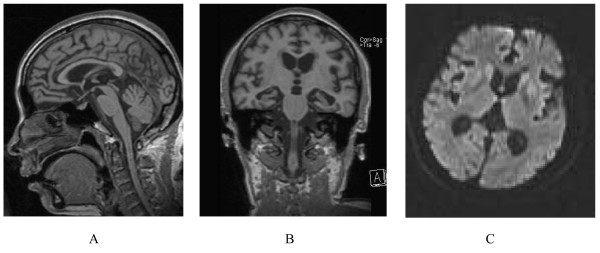 | |
| У человека с наследственной прионной болезнью наблюдается атрофия мозжечка. Это очень типично для GSS. | |
| Специальность | Неврология |
| Симптомы | трудности в разговоре, развитие слабоумие, потеря памяти, потеря зрения. |
| Причины | прион |
| Прогноз | (В СРЕДНЕМ) 5-6 лет с момента постановки диагноза |
Синдром Герстмана – Штройсслера – Шейнкера (GSS) является чрезвычайно редким, обычно семейным, смертельным нейродегенеративным болезнь которым страдают пациенты в возрасте от 20 до 60 лет. Он передается исключительно по наследству и встречается только в нескольких семьях по всему миру (согласно NINDS). Однако он относится к категории трансмиссивные губчатые энцефалопатии (TSE) из-за причинной роли, которую играет PRNP, человек прион белок.[1] Впервые о GSS сообщили австрийские врачи. Йозеф Герстманн, Эрнст Штраусслер и Илья Шейнкер в 1936 г.[2][3]
Семейные случаи связаны с аутосомный -доминантное наследование.[4]
Некоторые симптомы являются общими для GSS, например, прогрессирующий атаксия, пирамидальные знаки, и даже у взрослых слабоумие; они прогрессируют больше по мере прогрессирования болезни.[5]
Симптомы
Симптомы начинаются с медленно развивающихся дизартрия (затрудненная речь) и мозжечок туловищная атаксия (неустойчивость), а затем прогрессивный слабоумие становится более очевидным. Потеря памяти может быть первым признаком GSS.[6] Могут возникать экстрапирамидные и пирамидные симптомы и признаки, и болезнь может имитировать спиноцеребеллярную атаксию на начальной стадии. Миоклонус (спазматическое сокращение мышц) встречается реже, чем при Болезнь Крейтцфельдта-Якоба. У многих пациентов также наблюдается нистагм (непроизвольное движение глаз), нарушения зрения и даже слепота или глухота.[7] Невропатологические находки GSS включают широко распространенное отложение амилоидных бляшек, состоящих из аномально свернутого прионного белка.[6]
Выделяются четыре клинических фенотипа: типичный GSS, GSS с арефлексией и парестезией, GSS чистой деменции и GSS, подобный болезни Крейтцфельда-Якоба.[8]
Причины
GSS является одним из небольшое количество болезней которые вызываются прионами, классом патогенных белков, обладающих высокой устойчивостью к протеазы.[нужна цитата ]
Изменение в кодон 102 с пролин к лейцин был найден в прионный белок ген (PRNP, на хромосома 20 ) наиболее пострадавших людей.[9] Следовательно, кажется, что это генетический изменение обычно требуется для развития болезни.[нужна цитата ]
Диагностика
GSS можно определить с помощью генетического тестирования.[7] Тестирование на GSS включает исследование крови и ДНК, чтобы попытаться обнаружить мутировавший ген в определенных кодонах. Если генетическая мутация присутствует, пациент в конечном итоге будет поражен GSS, и из-за генетической природы заболевания потомство пациента предрасположено к более высокому риску наследования мутации.[нужна цитата ]
Уход
Нет лекарства от GSS, равно как и нет известного лечения, замедляющего прогрессирование болезни. Однако методы лечения и лекарства направлены на лечение или замедление эффектов симптомов. Их цель - попытаться как можно больше улучшить качество жизни пациента. Единственное лекарство, которое, похоже, может остановить болезнь, проходит испытания в Медицинском исследовательском центре в Лондоне. Это моноклональное антитело PRN100.[10]
Прогноз
Продолжительность болезни может составлять от трех месяцев до 13 лет, в среднем от пяти до шести лет.[6]
Исследование
GSS встречается очень редко, поэтому в его истории трудно точно отследить, откуда он произошел. В 1989 г. первая мутация гена прионного белка была идентифицирована в семье GSS (Elsevier Science, 2002). Прионные заболевания (трансмиссивные губчатые энцефалопатии) - дегенеративные заболевания головного мозга, которые, как считается, вызываются белком, который превращается в аномальную форму, называемую прионом (Gambetti Pierluigi, 2013). Позже выяснилось, что GSS имеет много разных типов мутаций генов, при этом некоторые сначала проявляют разные симптомы или имеют другие симптомы хуже, чем другие. Врачи в разных частях света выявляют все больше поколений и семей, у которых есть мутация. GSS трудно обнаружить по двум основным причинам: (1) заболевание зарегистрировано лишь в нескольких странах; и (2) данные о заболевании могут быть занижены из-за его клинического сходства с другими заболеваниями (Ghetti B, et al., 2003). Сородичи Индианы - самые крупные, насчитывающие более 8 поколений, и включают более 3000 человек, 57 из которых, как известно, затронуты (Б. Гетти и др., 1996).
Примечания
- ^ Либерски, Павел П. (2012). «Болезнь Герстмана – Штройсслера – Шейнкера». Достижения экспериментальной медицины и биологии. 724: 128–137. Дои:10.1007/978-1-4614-0653-2_10. ISSN 0065-2598. PMID 22411239.
- ^ синд / 2269 в Кто это назвал?
- ^ Gerstmann, J .; Sträussler, E .; Шейнкер, И. (1936). "Über eine eigenartige hereditär-familiäre Erkrankung des Zentralnervensystems. Zugleich ein Beitrag zur Frage des vorzeitigen lokalen Alterns". Zeitschrift für die gesamte Neurologie und Psychiatrie. 154: 736–762. Дои:10.1007 / bf02865827.
- ^ Де Микеле Дж., Поккиари М., Петрароли Р. и др. (Август 2003 г.). «Вариабельный фенотип в итальянской семье P102L Герстманна – Штройсслера – Шейнкера». Может J Neurol Sci. 30 (3): 233–6. Дои:10.1017 / S0317167100002651. PMID 12945948. Архивировано из оригинал на 28 января 2013 г.
- ^ Фарлоу, М.Р .; и другие. (1989). «Болезнь Герстмана-Штройсслера-Шейнкера. 1. Расширение клинического спектра». Неврология. 39 (11): 1446–1452. Дои:10.1212 / wnl.39.11.1446. PMID 2812321.
- ^ а б c Коллинз С., Маклин, Калифорния, Masters CL (сентябрь 2001 г.). «Синдром Герстмана – Штройсслера – Шейнкера, фатальная семейная бессонница и куру: обзор этих менее распространенных инфекционных губчатых энцефалопатий человека». J Clin Neurosci. 8 (5): 387–97. Дои:10.1054 / jocn.2001.0919. PMID 11535002.
- ^ а б Гамбетти, Пьерлуиджи. «Болезнь Герстмана – Штройсслера – Шейнкера». Руководства Merck: Медицинская онлайн-библиотека. Архивировано из оригинал 22 февраля 2011 г.. Получено 6 апреля, 2011.
- ^ Tesar A, Matej R, Kukal J, Johanidesova S, Rektorova I, Vyhnalek M, Keller J, Eliasova I, Parobkova E, Smetakova M, Musova Z, Rusina R (2019) Клиническая вариабельность синдрома Герстмана-Штреусслера-Шейнкера P102L. Энн Нейрол
- ^ Арата Х., Такашима Х., Хирано Р. и др. (Июнь 2006 г.). «Ранние клинические признаки и результаты визуализации при синдроме Герстмана – Штройсслера – Шейнкера (Pro102Leu)». Неврология. 66 (11): 1672–8. Дои:10.1212 / 01.wnl.0000218211.85675.18. PMID 16769939.
- ^ "ПРИОННЫЕ ЗАБОЛЕВАНИЯ И PRN100 "
внешняя ссылка
- Синдром Герстмана – Штройсслера – Шейнкера, MedicineNet.com
| Классификация |
|---|