Реакция Михаэлиса – Арбузова - Michaelis–Arbuzov reaction
| Реакция Михаэлиса – Арбузова | |
|---|---|
| Названный в честь | Август Михаэлис Александр Арбузов |
| Тип реакции | Реакция сцепления |
| Идентификаторы | |
| Портал органической химии | Арбузов-реакция |
| RSC ID онтологии | RXNO: 0000060 |
В Реакция Михаэлиса – Арбузова (также называемый Арбузов реакция) это химическая реакция из трехвалентный сложный эфир фосфора с алкилгалогенид сформировать пятивалентный разновидности фосфора и другой галогенид алкила. На рисунке ниже показаны наиболее распространенные типы субстратов, подвергающихся реакции Арбузова; сложные эфиры фосфита (1) реагировать на форму фосфонаты (2), фосфониты (3) реагировать на форму фосфинаты (4) и фосфиниты (5) реагировать на форму оксиды фосфина (6).
Реакция была обнаружена Август Михаэлис в 1898 г.,[1] и подробно исследован Александр Арбузов вскоре после этого.[2][3] Эта реакция широко используется для синтеза различных фосфонатов, фосфинаты, и оксиды фосфина. Опубликовано несколько обзоров.[4][5] Реакция также происходит для координированных фосфитных лигандов, как показано деметилированием {(C5ЧАС5) Co [(CH3O)3П]3}2+ дать {(C5ЧАС5) Co [(CH3O)2PO]3}−, который называется Клауи лиганд.
Механизм реакции
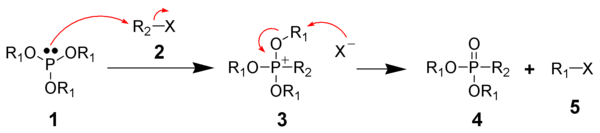
Реакция Михаэлиса – Арбузова инициируется SN2 атака из нуклеофильный виды фосфора (1 - фосфит) с электрофильный алкилгалогенид (2) дать фосфониевая соль как промежуточный (3). Эти промежуточные соединения иногда бывают достаточно стабильными для выделения, например, для триарилфосфитов, которые не вступают в реакцию с образованием фосфоната без термического расщепления промежуточного соединения (200 ° C) или расщепления спиртами или основаниями. Перемещенные галогенид анион тогда обычно реагирует через другой SN2 реакция на один из R1 углерода, замещая атом кислорода, чтобы дать желаемый фосфонат (4) и другой алкилгалогенид (5). Это подтверждается наблюдением, что киральный R1 группы испытывают инверсию конфигурации в углеродном центре, атакованном галогенид-анионом. Это то, что ожидается от SN2 реакция.[6] Также существуют доказательства карбокатион основанный на механизме деалкилирования аналогичный SN1 реакция, где R1 группа сначала диссоциирует от соли фосфония с последующей атакой аниона.[5] Сложные эфиры фосфита с третичными алкилгалогенидными группами могут вступать в реакцию, что было бы неожиданно, если бы только SN2 механизма работал. Дальнейшая поддержка этого SNМеханизм 1 типа связан с использованием реакции Арбузова в синтезе неопентил галогениды, класс соединений, которые заведомо инертны по отношению к SN2 реакции. По принципу микроскопическая обратимость, инертность неопентилгалогенидов по отношению к SN2 означает, что SN2 вряд ли может быть механизмом синтеза неопентилгалогенидов в этой реакции. Субстраты, которые не могут реагировать через SN2 путь или SN1 путь обычно не реагирует, в том числе винил и арил группы. Например, упомянутые выше триарилфосфиты обычно не вступают в реакцию, поскольку они образуют стабильные соли фосфония. Поскольку арильные группы не претерпевают SN1 и SN2 типа механизмов, триарилфосфиты лишены низкоэнергетического пути разложения фосфониевой соли. An аллильная перегруппировка механизм (SN2`) также был замешан в аллил и пропаргил галогениды.
Стереохимические эксперименты на циклических фосфитах выявили присутствие как пятивалентных фосфораны и промежуточные соединения четырехвалентного фосфония в химическое равновесие участие в стадии деалкилирования реакции с использованием 31P ЯМР. Разложение этих промежуточных продуктов в первую очередь обусловлено нуклеофильность аниона. Существует множество примеров, когда промежуточные соли фосфония достаточно стабильны, чтобы их можно было выделить, когда анион является слабонуклеофильным, например, с тетрафторборат или же тройной анионы.
Объем
Алкилгалогенид[5]
В качестве общего руководства реакционная способность органического галогенидного компонента может быть указана следующим образом: (от наиболее реакционной до наименее реактивной)

и

Обычно третичные алкилгалогениды, арилгалогениды и винилгалогениды не вступают в реакцию. Из этой тенденции есть заметные исключения, в том числе 1,2-дихлорэтен и тритил галогениды. Некоторые активированные арилгалогениды, часто содержащие гетероциклы были известны реакции. Йодобензол и замещенные производные, как известно, вступают в реакцию в условиях фотолиза. Вторичные алкилгалогениды часто плохо реагируют, образуя алкены как побочные продукты. Аллил- и пропаргилгалогениды также реакционноспособны, но могут проходить через SN2 или SN2` механизм. Реакция с первичными алкилгалогенидами и ацилгалогениды в целом все идет гладко. Тетрахлорметан что интересно, только один раз подвергается реакции с хлороформ инертен к условиям реакции. Когда атом галогенида находится в цепи сложного эфира от атома фосфора, изомеризация к соответствующему продукту Арбузова был известен без добавления галогенида алкила.
В Реакция Перкова является конкурирующим путем реакции для α-бром- и α-хлоркетонов. В условиях реакции образуется смесь продукта Perkow и обычного продукта Arbuzov, обычно в значительной степени благоприятствующая продукту Perkow. Использование более высоких температур во время реакции может привести к предпочтению продукта Арбузова. Реакция α-йодокетонов дает только продукт Арбузова.[7] Были разработаны другие способы получения β-кетофосфонатов.[8]
Реакция соединений трехвалентного фосфора с алкилфторидами ненормальна. Один из примеров такой реактивности показан ниже.

Реагент фосфора[5]
Общий вид реагента трехвалентного фосфора можно рассматривать следующим образом: причем A и B обычно представляют собой алкильные, алкокси или арилокси группы. Отвод электронов Известно, что группы замедляют скорость реакции, а электронодонорные группы увеличивают скорость реакции. Это согласуется с первоначальным воздействием фосфорного реагента на алкилгалогенид, поскольку этап определения ставки реакции. Реакция протекает гладко, когда группа R является алифатической. Когда все A, B и R представляют собой арильные группы, образуется стабильная соль фосфония, и реакция больше не протекает при нормальных условиях. Известно, что нагревание до более высоких температур в присутствии спиртов дает продукт изомеризации. Циклические фосфиты обычно реагируют с выделением нециклической группы OR, хотя для некоторых 5-членных колец требуется дополнительный нагрев для получения конечного циклического продукта.


Соли фосфита (Пример: R = Na) также могут вступать в реакцию с осаждением соответствующей соли галогенида натрия. Амидофосфиты и силилоксифосфиты использовались ранее для получения амидофосфонатов и фосфиновых кислот.

Перегруппировка типа Арбузова также может происходить, когда O из группы OR действует как уходящая группа в исходной SN2 атака фосфора. Известно, что это происходит только тогда, когда A и B представляют собой Cl.

Сложные эфиры фосфита - наименее реакционноспособный класс реагентов, используемых в этой реакции. Они реагируют с образованием фосфонатов. Для прохождения реакции им требуется максимальный нагрев (обычно 120–160 ° C). Эта высокая температура позволяет использовать фракционную перегонку для удаления полученного алкилгалогенида, хотя также можно использовать избыток исходного алкилгалогенида. Растворители часто не используются для этой реакции, хотя есть прецеденты улучшения селективности с ее использованием.
Фосфониты обычно более реакционноспособны, чем сложные эфиры фосфита. Они реагируют с образованием фосфинатов. Для реакции также требуется нагрев, но пиролиз превращение эфира в кислоту является обычной побочной реакцией. Плохая доступность замещенных фосфонитов ограничивает использование этого класса реагентов в реакции Арбузова. Гидрокси, тиол, карбоновая кислота, первичный и вторичный амин функциональные группы не могут использоваться с фосфонитами в реакции, поскольку все они реагируют с фосфонитом.
Фосфиниты - наиболее реактивный класс реагентов, используемых в этой реакции. Они реагируют с образованием оксидов фосфина. Они часто требуют очень небольшого нагревания (45 ° C) для протекания реакции и, как известно, самоизомеризуются без присутствия алкилгалогенидов.
Смотрите также
Рекомендации
- ^ Michaelis, A .; Kaehne, R. (1898). "Ueber das Verhalten der Jodalkyle gegen die sogen. Phosphorigsäureester or O-Phosphine". Берихте. 31: 1048–1055. Дои:10.1002 / cber.189803101190.
- ^ Арбузов, А. Э. (1906). J. Russ. Phys. Chem. Soc. 38: 687. Отсутствует или пусто
| название =(помощь) - ^ Арбузов, А. Э. (1906). Chem. Центр. II: 1639. Отсутствует или пусто
| название =(помощь) - ^ Арбузов, Б.А. (1964). «Михаэлис-Арбусов- унд Перков-Реактионен». Pure Appl. Chem. 9 (2): 307–353. Дои:10.1351 / pac196409020307. S2CID 93719226.
- ^ а б c d Bhattacharya, A.K .; Тьягараджан, Г. (1981). «Перестановка Михаэлиса – Арбузова». Chem. Ред. 81 (4): 415–430. Дои:10.1021 / cr00044a004.
- ^ Gerrard, W .; Грин, У. Дж. (1951). «568. Механизм образования диалкилалкилфосфонатов». J. Chem. Soc.: 2550. Дои:10.1039 / младший 9510002550.
- ^ Jacobsen, H.I .; Гриффин, М. Дж .; Preis, S .; Дженсен, Э. В. (1957). «Фосфоновые кислоты. IV. Получение и реакции β-кетофосфонатных и енольных фосфатных эфиров». Варенье. Chem. Soc. 79 (10): 2608. Дои:10.1021 / ja01567a067.
- ^ Nagata, W .; Wakabayashi, T .; Хаясе, Ю. (1988). «Диэтил 2- (циклогексиламино) винилфосфонат». Органический синтез.; Коллективный объем, 6, п. 448
внешняя ссылка
- Ford-Moore, A.H .; Перри, Б. Дж. Органический синтез, Сб. Vol. 4, стр. 325 (1963); Vol. 31, стр. 33 (1951). (Статья )
- Davidsen, S.K .; Phllips, G.W .; Мартин, С.Ф. Органический синтез, Сб. Vol. 8, стр. 451 (1993); Vol. 65, стр. 119 (1987). (Статья )
- Эндерс, Д .; фон Берг, S .; Джанделейт, Б. Органический синтез, Сб. Vol. 10, стр. 289 (2004); Vol. 78, стр. 169 (2002). (Статья )