Реакции медьорганических реагентов - Reactions of organocopper reagents
Реакции медьорганических реагентов вовлекать разновидность содержащие медь-углеродные связи, действующие как нуклеофилы в присутствии органических электрофилы. Медьорганические реагенты теперь широко используются в органический синтез как мягкие, селективные нуклеофилы для реакций замещения и конъюгированного присоединения.[1]
С момента открытия, что галогениды меди (I) катализировать сопряженное добавление Реактивы Гриньяра в 1941 г.,[2] Медьорганические реагенты возникли как слабоосновные, нуклеофильные реагенты для реакций замещения и присоединения. Состав медьорганических соединений зависит от метода их получения, и различные виды медьорганических реагентов проявляют разные свойства. профили реактивности. В результате диапазон реакций с участием медьорганических реагентов чрезвычайно широк.
- Медноорганические комплексы (RCu) образуются при объединении галогенида меди (I) и литийорганического соединения. В сочетании с Льюис кислый добавки, такие как трифторид бора эфират, эти реагенты используются для реакций присоединения конъюгатов.[3]
- Купраты низшего порядка (Р2CuLi, также известный как Реактивы Гилмана ) результат обработки медьорганических комплексов эквивалентом литийорганический. Альтернативно, они могут быть образованы обработкой галогенида меди (I) двумя эквивалентами литийорганического соединения. Они претерпевают реакции замещения, сопряженного присоединения и карбокупрации в присутствии соответствующих органических субстратов.[4] Смешанные реагенты Гилмана состоят из двух разных R-групп, одна из которых обычно является непередаваемой «фиктивной» группой.
- Цианокупраты низшего порядка (RCu (CN) Li) аналогичным образом получают из литийорганического соединения и цианид меди (I); однако промежуточные медноорганические комплексы не образуются во время этой реакции, и поэтому необходим только один эквивалент литийорганического реагента.[1] Цианокупраты подвергаются SN2 'замещение в присутствии аллильных электрофилов и реакции сопряженного присоединения в присутствии Enones.
- Цианокупраты высшего порядка (Р2Cu (CN) Li2) образуются при взаимодействии двух эквивалентов литийорганического соединения с цианид меди (I). Эти реагенты более реактивны по отношению к замещению, чем соответствующие цианокупраты более низкого порядка.[5]
Механизм и стереохимия
Реакции замещения
Механизм нуклеофильного замещения органокупратами низшего порядка во многом зависит от структуры субстрата, органокупрата и условий реакции. Ранние данные предполагали, что прямая SN2 смещения происходили;[6] однако более поздние результаты предполагают, что имеет место обратное окислительное присоединение меди (I) к связи уходящей углеродной группы с образованием промежуточного соединения меди (III), которое затем подвергается восстановительному отщеплению с образованием связанного продукта.[7] Оба эти механизма предсказывают инверсию электрофильного углерода, которая наблюдается в ряде случаев.[8] С другой стороны, эксперименты с радикальными ловушками и наблюдение рацемизация во время замещения предполагают радикальный механизм.[9]
(1)

Реакции сопряженного добавления
В 1941 году Хараш обнаружил, что Реактивы Гриньяра добавляют к циклогексенону в присутствии Cu (I), что приводит к 1,4-присоединению вместо 1,2-присоединения.[10] Эта работа предвещала обширные исследования сопряженные добавки к Enones с органокупратами. Обратите внимание, что если Реактив Гриньяра (например, RMgBr), реакция с еноном вместо этого будет протекать через 1,2-присоединение. Механизм 1,4-присоединения купратов к енонам заключается в нуклеофильном присоединении разновидностей Cu (I) к бета-углероду алкена с образованием промежуточного соединения Cu (III) с последующим восстановительным отщеплением Cu (I).[11] В исходной статье, описывающей эту реакцию, метилмагнийбромид реагирует с изофорон с добавлением 1 мольного процента и без него хлорид меди (I) (см. рисунок).[10]
![Добавление метилмагнийбромида к изофорону. [10]](http://upload.wikimedia.org/wikipedia/commons/thumb/9/97/Coppercatalyzedenonegrignardaddition.png/400px-Coppercatalyzedenonegrignardaddition.png)
Без добавления соли основные продукты алкоголь B (42%) из нуклеофильное присоединение к карбонильной группе и диен C (48%) как его реакция дегидратации товар. С добавлением соли основным продуктом является 1,4-аддукт. А (82%) с некоторыми C (7%).
Также возможно 1,6-добавление, например, на одной стадии промышленного производства фулвестрант:[12]

Энантиоселективные варианты
Реакции диастереоселективного присоединения конъюгатов хиральных органокупратов обеспечивают β-функционализированные кетоны с высоким выходом и диастереоселективностью. Недостатком этих реакций является необходимость полного эквивалента энантиочистка исходный материал.[13]
(3)

Совсем недавно были разработаны каталитические энантиоселективные методы, основанные на катализируемом медью (I) сопряженном присоединении реакций Гриньяра к енонам. Предлагаемый механизм включает трансметалляцию из реактива Гриньяра в медь, добавление конъюгата и определяющее скорость восстановительное удаление (см. Аналогичный верхний путь в уравнении (2)).[14]
(4)

Каталитические реакции
Виниловый и арильный реагенты Гриньяра связываются с первичными алкилгалогенидами в присутствии каталитического количества галогенидной соли меди (I). Использование Ли2CuCl4 Вместо простых солей галогенида меди (I) (CuX) выход этих реакций сочетания улучшается.[15]
(5)

Добавление реактивов Гриньяра к алкинам облегчается каталитическим количеством галогенида меди. Трансметалляция до меди и карбокупрация сопровождаются трансметаллированием продукта. алкен вернуться к магний. Дополнение син если координирующая группа не находится поблизости в субстрате, в этом случае добавление становится анти и урожайность улучшается.[16]
(6)

Стехиометрические реакции
Пропаргилметансульфинаты являются полезными субстратами для синтеза аллены из стехиометрических медноорганических комплексов. В этом случае комплексы образовывались на месте через комбинацию реактива Гриньяра, бромида меди (I) и бромида лития. Медноорганические комплексы очень часто требуют активации кислотой Льюиса для эффективной реакции; бромид магния генерируется на месте в этом случае служит активирующей кислотой Льюиса.[17]
(7)

Комплексы алкенил-меди, легко образующиеся посредством карбокупрации, полезны для введения винильной группы в β-положение карбонильного соединения. В этом случае, как указано выше, бромид магния служит в качестве активирующей кислоты Льюиса.[18]
(8)

Эпоксид открытие органокупратов очень избирательно для менее затрудненного положения. Замещение происходит при полной инверсии конфигурации электрофильного углерода.[19]
(9)

Обычно органокупраты реагируют с аллильными электрофилами в анти SN2 мода. В приведенной ниже реакции наблюдалась почти полная инверсия конфигурации, несмотря на присутствие второго стереоцентра в кольце.[20]
(10)

Сопряженное добавление органокупратов широко используется в органическом синтезе. Купраты винилового эфира служат в качестве удобных ацильный анион эквиваленты в реакциях сопряженного присоединения к енонам. Полученные простые эфиры енола могут быть гидролизованы до 1,4-дикетонов, которые трудно получить с помощью традиционной химии карбонила.[21]
(11)

Использование добавок в сочетании со стехиометрическим количеством медноорганических комплексов увеличивает скорость и выход многих реакций. В частности, медноорганические комплексы медленно реагируют в отсутствие кислоты Льюиса. Хотя бромид магния генерирует на месте от реакции реактивов Гриньяра и галогенидов меди (I) эту роль могут выполнять (см. выше), также могут использоваться внешние кислоты Льюиса. В присутствии эфирата трифторида бора медноорганические комплексы могут добавляться к стерически перегруженным енонам с умеренным выходом (осуществление такого же преобразования с органокупратом было бы затруднительно).[22]
(12)
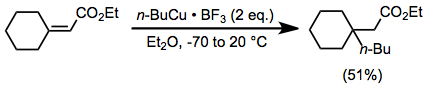
Эфират трифторида бора также полезен в качестве добавки в реакциях цианкупратов более высокого порядка. Использование 2-тиенильной группы в качестве «фиктивного» заместителя в цианокупрате позволяет сохранить потенциально ценный литийорганический реагент, используемый для образования цианкупрата (поскольку в медьсодержащих побочных продуктах присутствует только фиктивная группа). В отсутствие эфирата трифторида бора в этом случае реакции не наблюдали.[23]
(13)
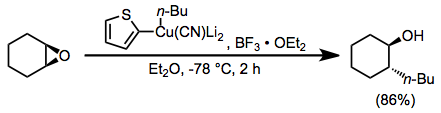
Реакции присоединения конъюгатов цианокупратов высшего порядка представляют собой еще одно полезное применение эфирата трифторида бора. Виниловая группа в этой реакции переносится избирательно; это отличается от реакций замещения с использованием того же реагента, которые приводят к селективному переносу метильной группы.[24]
(14)

Алкилирование аминов
Вторичные амины можно алкилировать купратами. Реакция основана на окислительном связывании амида лития-алкилмеди, который, как сообщается, образуется in situ во время реакции между диалкилкупратами лития и первичными или вторичными амидами.[25]

Синтетические приложения
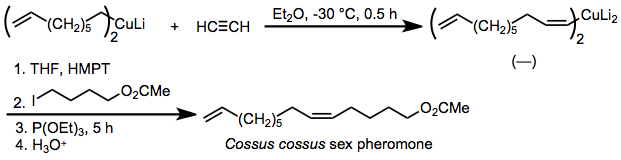
Поскольку стереоселективность карбокупрации чрезвычайно высока, эта реакция была применена для синтеза феромоны в котором геометрическая чистота двойные связи имеет решающее значение. Одним из примеров является феромон насекомых Cossus Cossus, который синтезируется син-селективная карбокупрация ацетилен и алкилирование полученного органокупрата в присутствии добавленного фосфита.[26]
(15)
Рекомендации
- ^ а б Lipshutz, B.H .; Сенгупта, С. Орг. Реагировать. 1992, 41, 135. Дои:10.1002 / 0471264180.or041.02
- ^ Kharasch, M. S .; Тоуни, П. О. Варенье. Chem. Soc. 1941, 63, 2308.
- ^ Кансал, В. К .; Тейлор, Р. Дж. К. J. Chem. Soc. Perkin Trans. 1 1984, 703.
- ^ Познер, Г. Х. Орг. Реагировать. 1975, 22, 253.
- ^ Lipshutz, B.H .; Wilhelm, R. S .; Флойд, Д. М. Варенье. Chem. Soc. 1981, 103, 7672.
- ^ Тамура, М .; Кочи, Дж. К. J. Organomet. Chem. 1972, 42, 205.
- ^ Кори, Э. Дж .; Боаз, Н.В. Tetrahedron Lett. 1984, 25, 3059.
- ^ Johnson, C. R .; Дутра, Г.А. Варенье. Chem. Soc. 1973, 95, 7777.
- ^ Ashby, E.C .; Коулман, Д. J. Org. Chem. 1987, 52, 4554.
- ^ а б c Kharasch, M. S .; Тоуни, П. О. (1941). «Факторы, определяющие протекание и механизмы реакций Гриньяра. II. Влияние металлических соединений на реакцию между изофороном и метилмагнием бромидом». Журнал Американского химического общества. 63 (9): 2308–2316. Дои:10.1021 / ja01854a005. ISSN 0002-7863.
- ^ Накамура, Эйити; Мори, Сейджи (2000). «Зачем ты медь? Структуры и механизмы реакции кластеров купратов в органической химии». Angewandte Chemie. 39 (21): 3750–3771. Дои:10.1002 / 1521-3773 (20001103) 39:21 <3750 :: AID-ANIE3750> 3.0.CO; 2-L. PMID 11091452.
- ^ Фулвестрант: от лаборатории к производству в промышленных масштабах Ева Дж. Брейзер, Филип Дж. Хоган, Чиу В. Люнг, Энн О'Керни-Макмаллан, Элисон К. Нортон, Лин Пауэлл, Грэм Э. Робинсон и Эмир Г. Уильямс Исследования и разработки органических процессов 2010, 14, 544 –552 Дои:10.1021 / op900315j
- ^ Malmberg, H .; Nilsson, M .; Уллениус, К. Tetrahedron Lett. 1982, 23, 3823.
- ^ Арутюнян, С .; Лопес, Ф .; Браун, В .; Correa, A .; Peña, D .; Badorrey, R .; Meetsma, A .; Minnaard, A .; Феринга, Б.Л. Варенье. Chem. Soc. 2006, 128, 9103.
- ^ Нуномото, S .; Kawakami, Y .; Ямасита, Ю. J. Org. Chem. 1983, 48, 1912.
- ^ Жуссом, B. Ph.D. Диссертация, Университет Бордо, Франция, 1977 г.
- ^ Kleijn, H .; Elsevier, C.J .; Westmijze, H .; Meijer, J .; Вермеер, П. Tetrahedron Lett. 1979, 3101.
- ^ Марфат, А .; McGuirk, P.R .; Хелквист, П. J. Org. Chem. 1979, 44, 3888.
- ^ Johnson, M. R .; Наката, Т .; Киши, Y. Tetrahedron Lett. 1979, 4343.
- ^ Геринг, Х.Л .; Кантнер, С.С. J. Org. Chem. 1981, 46, 2144.
- ^ Boeckman, R.K .; Рамая, М. J. Org. Chem. 1977, 42, 1581.
- ^ Yamamoto, Y .; Ямамото, S .; Yatagai, S .; Ishihara, Y .; Маруяма, К. J. Org. Chem. 1982, 47, 119.
- ^ Lipshutz, B.H .; Паркер, Д. А .; Козловски, Дж. А .; Нгуен, С.Л. Tetrahedron Lett. 1984, 25, 5959.
- ^ Lipshutz, B.H .; Wilhelm, R. S .; Козловский, Ю.А. J. Org. Chem. 1984, 49, 3938.
- ^ Yamamoto, H .; Марука, К. (1980). «Новое N-алкилирование аминов медьорганическими реагентами». J. Org. Chem. 45: 2739–2740. Дои:10.1021 / jo01301a048.
- ^ Cahiez, G .; Алексакис, А .; Норман, Дж. Ф. Tetrahedron Lett. 1978, 2027.