Мечение подложек изотопными концевыми аминогруппами - Terminal amine isotopic labeling of substrates
Мечение подложек изотопными концевыми аминогруппами (ХВОСТ) - метод в количественная протеомика что определяет белок содержание образцов на основе N-концевой фрагменты каждого белка (N-концевой пептиды ) и обнаруживает различия в содержании белка между образцами.
Как и другие методы, основанные на N-концевых пептидах, в этом анализе используются трипсин для разделения белков на фрагменты и отделения N-концевых пептидов (фрагментов, содержащих N-концы исходных белков) от других фрагментов (внутренних триптических пептидов). TAILS изолирует N-концевые пептиды путем идентификации и удаления внутренних триптических пептидов. Этот отрицательный выбор позволяет методу TAILS обнаруживать все N-концы в данных образцах. Альтернативные методы, которые полагаются на свободную аминогруппу N-конца для идентификации N-концевых пептидов, не могут обнаружить некоторые N-концы, потому что они «естественным образом заблокированы» (т.е. природный белок не имеет свободной аминогруппы).
Метод ХВОСТОВ имеет ряд применений, включая идентификацию новых субстраты и протеазы (включая те, которые имеют неизвестную и широкую специфику)[1] и как способ определения концов белков, обеспечивающий аннотацию белков. TAILS также можно использовать для связывания протеаз с множеством определенных биологических путей при таких заболеваниях, как рак, чтобы получить более четкое представление о субстратах и протеазах, участвующих в болезненном состоянии.[2]
Метод
TAILS - это двух- или трехмерный протеомный анализ для мечения и выделения N-концевых пептиды, разработанная группой в Университет Британской Колумбии.[1] Метод TAILS разработан для сравнения клеток, обработанных множеством протеаз, и контрольных протеомных клеток.[2] Образцы могут быть получены из различных источников, включая ткани, фибробласты, раковые клетки и жидкие выделения.
Этот анализ выделяет N-концевые пептиды путем удаления внутренних триптических пептидов через ультрафильтрация оставляя меченые зрелые N-концевые и нео-N-концевые пептиды для анализа тандемом масс-спектрометрии (МС / МС). Этот отрицательный выбор позволяет методу TAILS обнаруживать все N-концы в данных образцах. Альтернативные методы, которые полагаются на свободную аминогруппу N-конца для выделения N-концевых пептидов, не могут обнаружить естественно заблокированные N-концы, поскольку они не имеют свободной аминогруппы.
Для экспериментов TAILS требуется только небольшой образец пептида (100–300 мкг), его можно использовать с протеазами с неизвестной или широкой специфичностью и поддерживает различные методы мечения образцов. Однако он идентифицирует ~ 50% белков по двум или более различным и уникальным пептидам (один из исходных зрелых N-концевых и / или один или несколько нео-N-концевых пептидов через сайт расщепления), которые не представляют независимых биологических событий, таким образом не может быть усреднен для количественной оценки[требуется разъяснение ]. У него также есть трудности с проверкой результатов для анализа N-концевого соединения одного пептида.[требуется разъяснение ].[1]
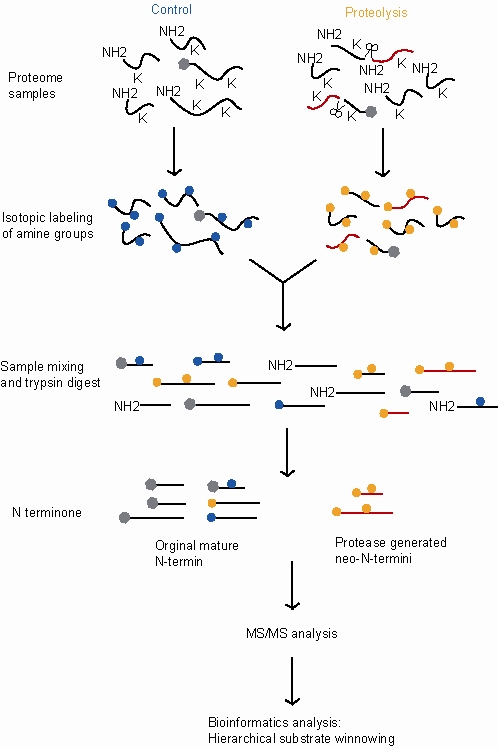
Следующие шаги предназначены для анализа диметилирования-TAILS, сравнивая контрольный образец (демонстрирующий нормальную протеолитическую активность) и обработанный образец (который в этом примере проявляет дополнительную протеолитическую активность).
- Всесторонний протеом протеолиз возникающие как в обработанных, так и в контрольных образцах с дополнительной протеолитической активностью в обработанном образце.
- Инактивация протеаз и денатурация и восстановление белков.
- Маркировка стабильными изотопами. Это позволяет отличать пептиды, происходящие из контрольного образца, от пептидов, происходящих из обработанного образца, и сравнивать их относительное количество. В этом примере мечение применяется восстановительным диметилированием первичных аминов с использованием либо тяжелого (d (2) C13) -формальдегида для обработанных образцов, либо легкого (d (O) C12) -формальдегида для контролей. Эта реакция катализируется цианоборгидридом натрия и присоединяет меченые метильные группы к лизинаминам и свободные (∝) - аминогруппы на N-концах белков и продуктов расщепления протеазой.
- Блокирование реактивных аминогрупп. Это позволяет идентифицировать внутренние триптические пептиды позже в процессе, поскольку они будут единственными пептидами с реактивными аминогруппами. В этом примере реакция мечения (восстановительное диметилирование) также блокирует реакционноспособные аминогруппы.
- Объединение. Теперь два меченых протеома смешаны. Это гарантирует, что образцы обрабатываются одинаково на всех последующих этапах, что позволяет более точно измерить относительные количества белков в двух образцах.
- Трипсинизация. Это разбивает каждый белок на фрагменты. Меченые N-концы исходных белков остаются заблокированными, тогда как новые внутренние триптические пептиды имеют свободный N-конец.
- Отрицательный выбор. Сверхразветвленный полиглицерин-альдегидный (HPG) полимер, специфичный для связывания триптических пептидов, добавляется к образцу и вступает в реакцию с вновь образованными триптическими пептидами через их свободные N-концы. Как и в шаге 3 выше, эта реакция катализируется цианоборгидридом натрия. Ацетилированные и меченные изотопами белковые пептиды диметилированного лизина и нео (новые) -N-концевые пептиды нереактивны и остаются несвязанными и могут быть отделены от поливнутренних триптических пептидных комплексов с использованием ультрафильтрация.
- В элюированный несвязанные белки имеют высокую концентрацию N-концевых пептидов и нео-N-концевых пептидов.
- Затем этот элюированный образец количественно определяется и анализ завершается МС / МС.
- Последний шаг в TAILS включает: биоинформатика. Использование иерархического процесса отсеивания субстратов, который отличает продукты протеолиза от фона и нерасщепленные белки количественным определением изотопов пептидов и определенными критериями биоинформатического поиска.[1][2]
Типы
Типы ХВОСТОВ различаются методами, используемыми для блокирования и мечения аминогрупп белков и продуктов расщепления протеазами. Эти аминогруппы включают лизин-амины и свободные () -аминогруппы на N-концах белков.
Процедура диметилирования-TAILS - это процедура, основанная на химической маркировке, которая выполняется в один этап с использованием изотопных реагентов, реагирующих с амином. Маркировка двух образцов использует либо 12CH2-формальдегид (свет) или 13CD2-формальдегид (тяжелый) и использует цианоборгидрид натрия в качестве катализатора.[1] Преимущество этого метода заключается в том, что он надежен, эффективен и рентабелен. Процедура маркировки контрольных образцов и образцов, обработанных протеазой, должна выполняться отдельно, прежде чем их можно будет объединить, и она ограничена двумя образцами на эксперимент, которые могут быть недостаток, если необходимо исследовать несколько образцов одновременно.[1]
Стабильный изотоп мечение аминокислотами в культуре клеток (СИЛАК) - это процедура, которую можно выполнить in vivo. Эту процедуру можно использовать во всех лабораториях по культивированию клеток, и она является стандартным методом маркировки. Этот метаболический маркировка позволяет ингибировать данную протеазу в биологических образцах и анализировать ex vivo обработка.[1] Преимущество использования этого метода метаболической маркировки перед химической маркировкой состоит в том, что он позволяет надежно, быстро и эффективно различать реальные белки клеточного происхождения, которые исследуются на предмет наличия примесей, таких как белки сыворотки. СИЛАЧНЫЙ ХВОСТ можно использовать для анализа до пяти мультиплекс образцы. SILAC не подходит для клинически значимых человеческих образцов, которые не могут быть метаболически помечены. SILAC - дорогостоящий метод, который может быть неприемлемым для большинства лабораторий.[1]
В изобарный тег для относительной и абсолютной количественной оценки (iTRAQ) или iTRAQ-TAILS позволяет проводить количественный анализ нескольких образцов одновременно. Этот метод позволяет одновременно анализировать от 4-8 образцов в мультиплексных экспериментах с использованием четырех- и восьмиплексных реагентов iTRAQ. Этот метод обеспечивает высокую точность идентификации и количественного определения. образцов и обеспечивает более воспроизводимый анализ дубликатов образцов.[1] Как и другие методы iTRAQ, iTRAQ-TAILS требует масс-спектрометра MALDI и дорогостоящих реагентов iTRAQ.
Альтернативные методы
Существует несколько альтернативных подходов к изучению N-концов и продуктов протеолиза.
Ацетилирование аминов с последующим триптическим расщеплением и биотинилированием свободных N-концевых пептидов использует химическое (ацетилирование) для мечения свободных лизинов и N-концов. Затем заблокированные N-концы выбираются отрицательно. Однако естественные свободные внутренние N-концы и заблокированные N-концы не могут быть различимы после ацетилирования. В этом методе не используется изотопное мечение, поэтому количественно оценить результаты сложно. Кроме того, трудно отличить экспериментальные продукты протеолиза от фоновых.[3]
Гуанидинирование лизина с последующим биотинилированием N-концов использует химические вещества для блокирования остатков лизина и метки свободных N-концов. Затем выбираются помеченные свободные N-концы. Обратной стороной этого метода является то, что результаты не могут быть применены к статистической модели с использованием нерасщепленных пептидов из-за невозможности захвата естественно заблокированных N-концов. Поскольку это не связано с изотопной маркировкой, результаты не могут быть определены количественно. Сайт расщепления также должен быть уже известен для мечения.[4]
Субтилигазное биотинилирование N-концов использует ферментативное мечение N-концевых пептидов, но не использует химические вещества, блокирующие лизин. Без блокировки лизина многие из расщепленных N-концевых пептидов будут слишком короткими для идентификации. Результаты могут сильно зависеть от свойств субтилигазы, поэтому могут быть смещены. Этот метод не захватывает естественно заблокированные N-концы, а также не использует изотопное мечение, поэтому было бы трудно количественно оценить результаты.[5]
ITRAQ-маркировка N-конца использует iTRAQ для маркировки N-конца. Нео-N-концевые пептиды выбирают in silico. Обратной стороной этого метода является то, что необходим масс-спектрометр MALDI, а необходимые реагенты iTRAQ являются дорогостоящими. Этот метод не захватывает естественно заблокированные N-концы. Для всего процесса потребуется от 50 до 100 мг пептидов.[6]
Комбинированная фракционная диагональная хроматография (COFRADIC) позволяет по-разному мечения для естественно заблокированных N-концов и нео-N-концов, генерируемых протеазой. Все заблокированные N-концы выбираются отрицательно. Однако этот процесс требует многих химических обработок, хроматографии и масс-спектрометрии. Наилучшие результаты разделения зависят от модификации аминокислоты, такой как окисление метионина, не происходящей во время работы. Этот метод требует 150 анализов МС / МС на образец, но образцы можно объединить для масс-спектрометрии (и количество анализов может быть уменьшено). Этот метод подходит для использования с протеазами с неизвестной или широкой специфичностью.[7]
Смотрите также
Рекомендации
- ^ а б c d е ж грамм час я Клейфельд, Одед; Дусе, Ален; Прудова, Анна; Ауф Дем Келлер, Ульрих; Джоя, Магда; Кижаккедату, Джаячандран Н; В целом, Christopher M (2011). «Идентификация и количественная оценка протеолитических событий и естественного N-конца путем мечения субстратов изотопным концевым амином». Протоколы природы. 6 (10): 1578–611. Дои:10.1038 / nprot.2011.382. PMID 21959240.
- ^ а б c Клейфельд, Одед; Дусе, Ален; Ауф Дем Келлер, Ульрих; Прудова, Анна; Шиллинг, Оливер; Kainthan, Rajesh K; Старр, Аманда Э; Фостер, Леонард Дж; и другие. (2010). «Изотопное мечение концевых аминов в сложных образцах позволяет идентифицировать N-концы белка и продукты расщепления протеазой». Природа Биотехнологии. 28 (3): 281–8. Дои:10.1038 / nbt.1611. PMID 20208520.
- ^ Макдональд, Люси; Робертсон, Дункан Х. Л.; Херст, Джейн Л; Бейнон, Роберт Дж (2005). «Позиционная протеомика: Селективное восстановление и анализ N-концевых протеолитических пептидов». Природные методы. 2 (12): 955–7. Дои:10.1038 / nmeth811. PMID 16299481.
- ^ Тиммер, Джон С .; Энокссон, Мари; Дикий Клык, Эрик; Чжу, Вэньхун; Игараси, Ёсинобу; Дено, Жан-Бенар; Ма, Юлянь; Даммит, Бенджамин; и другие. (2007). «Профилирование конститутивных протеолитических событий in vivo». Биохимический журнал. 407 (1): 41–8. Дои:10.1042 / BJ20070775. ЧВК 2267409. PMID 17650073.
- ^ Махрус, Сами; Тринидад, Джонатан К .; Баркан, Дэвид Т .; Сали, Андрей; Burlingame, Alma L .; Уэллс, Джеймс А. (2008). «Глобальное секвенирование сайтов протеолитического расщепления при апоптозе с помощью специфической маркировки белка N-конца». Клетка. 134 (5): 866–76. Дои:10.1016 / j.cell.2008.08.012. ЧВК 2566540. PMID 18722006.
- ^ Энокссон, Мари; Ли, Цзинвэй; Ivancic, Melanie M .; Тиммер, Джон С .; Дикий Клык, Эрик; Ерошкин Алексей; Salvesen, Guy S .; Тао, В. Энди (2007). «Идентификация протеолитических сайтов расщепления с помощью количественной протеомики». Журнал протеомных исследований. 6 (7): 2850–8. Дои:10.1021 / pr0701052. PMID 17547438.
- ^ Геваерт, Крис; Гетальс, Марк; Мартенс, Леннарт; Ван Дамм, Йозеф; Стаес, Ан; Thomas, Grégoire R .; Вандекеркхове, Жоэль (2003). «Изучение протеомов и анализ процессинга белков с помощью масс-спектрометрической идентификации отсортированных N-концевых пептидов». Природа Биотехнологии. 21 (5): 566–9. Дои:10.1038 / nbt810. PMID 12665801.