Байесовский вывод в филогении - Bayesian inference in phylogeny
| Классификация | Эволюционная биология |
|---|---|
| Подклассификация | Молекулярная филогенетика |
| Оптимально критерии поиска | Байесовский вывод |
Байесовский вывод филогении использует функцию правдоподобия для создания величины, называемой апостериорной вероятностью деревьев, используя модель эволюции, основанную на некоторых априорных вероятностях, создавая наиболее вероятное филогенетическое дерево для заданных данных. Байесовский подход стал популярным благодаря достижениям в скорости вычислений и интеграции Цепь Маркова Монте-Карло (MCMC) алгоритмы. Байесовский вывод имеет ряд приложений в молекулярная филогенетика и систематика.
Байесовский вывод о предпосылках и основаниях филогении

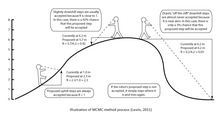
Байесовский вывод относится к вероятностному методу, разработанному преподобным Томасом Байесом на основе Теорема Байеса. Опубликованный посмертно в 1763 году, он был первым выражением обратной вероятности и основой байесовского вывода. Независимо, не зная о работе Байеса, Пьер-Симон Лаплас разработал теорему Байеса в 1774 году.[1]
Байесовский вывод широко использовался до 1900-х годов, когда произошел переход к частотному выводу, в основном из-за вычислительных ограничений. Основанный на теореме Байеса, байесовский подход объединяет априорную вероятность дерева P (A) с вероятностью данных (B) для получения апостериорного распределения вероятностей на деревьях P (A | B). Апостериорная вероятность дерева будет указывать на вероятность того, что дерево будет правильным, поскольку дерево с наивысшей апостериорной вероятностью выбрано для лучшего представления филогении. Это было введение Цепь Маркова Монте-Карло (MCMC) методы Николаса Метрополиса в 1953 году, которые произвели революцию в байесовском выводе и к 1990-м годам стали широко использоваться среди филогенетиков. Некоторые преимущества перед традиционными максимальная экономия и максимальная вероятность Методы - это возможность учета филогенетической неопределенности, использования априорной информации и включения сложных моделей эволюции, которые ограничивали вычислительный анализ традиционных методов. Несмотря на преодоление сложных аналитических операций, апостериорная вероятность все же включает суммирование по всем деревьям и для каждого дерева интегрирование по всем возможным комбинациям значений параметров модели замещения и длин ветвей.
Методы MCMC можно описать в три этапа: сначала с помощью стохастического механизма новое состояние для Цепь Маркова предлагается. Во-вторых, вычисляется вероятность того, что это новое состояние будет правильным. В-третьих, предлагается новая случайная величина (0,1). Если это новое значение меньше вероятности принятия, новое состояние принимается и состояние цепочки обновляется. Этот процесс выполняется тысячи или миллионы раз. Количество посещений одного дерева в ходе цепочки является всего лишь допустимым приближением его апостериорной вероятности. Некоторые из наиболее распространенных алгоритмов, используемых в методах MCMC, включают алгоритмы Metropolis-Hastings, Metropolis-Coupling MCMC (MC³) и LOCAL алгоритм Ларгета и Саймона.
Алгоритм Метрополиса-Гастингса
Одним из наиболее распространенных используемых методов MCMC является Алгоритм Метрополиса-Гастингса,[2] модифицированная версия оригинального алгоритма Метрополиса.[3] Это широко используемый метод случайной выборки из сложных и многомерных вероятностей распределения. Алгоритм Метрополиса описывается следующими шагами:[4]
- Исходное дерево, Tя, выбирается случайным образом
- Соседское дерево, Tj, выбирается из коллекции деревьев.
- Отношение R вероятностей (или функций плотности вероятности) Tj и тя вычисляется следующим образом: R = f (Tj) / f (Tя)
- Если R ≥ 1, Tj принимается как текущее дерево
- Если R <1, Tj принимается за текущее дерево с вероятностью R, иначе Tя хранится
- На этом этапе процесс повторяется с шага 2 N раз.
Алгоритм продолжает работу, пока не достигнет равновесного распределения. Также предполагается, что вероятность предложения нового дерева Tj когда мы находимся в состоянии старого дерева Tя, такая же вероятность предложить Tя когда мы находимся в Tj. Когда это не так, применяются поправки Гастингса. Цель алгоритма Метрополиса-Гастингса - создать набор состояний с определенным распределением до тех пор, пока марковский процесс не достигнет стационарного распределения. Алгоритм состоит из двух компонентов:
- Возможный переход из одного состояния в другое (i → j) с использованием функции вероятности перехода qя, j
- Движение цепи в состояние j с вероятностью αя, j и остается в i с вероятностью 1 - αя, j.[5]
MCMC для мегаполисов
Алгоритм MCMC, связанный с мегаполисом (MC³) [6] был предложен для решения практической проблемы перемещения цепи Маркова через пики, когда целевое распределение имеет несколько локальных пиков, разделенных низкими впадинами, которые, как известно, существуют в пространстве дерева. Это имеет место во время эвристического поиска в дереве в соответствии с критериями максимальной экономии (MP), максимального правдоподобия (ML) и минимального развития (ME), и то же самое можно ожидать для поиска в стохастическом дереве с использованием MCMC. Эта проблема приведет к неправильному приближению образцов к апостериорной плотности. (MC³) улучшает перемешивание цепей Маркова при наличии нескольких локальных пиков в апостериорной плотности. Он запускает несколько (m) цепочек параллельно, каждая для n итераций и с разными стационарными распределениями. , , где первый, - целевая плотность, а , выбраны для улучшения перемешивания. Например, можно выбрать пошаговый нагрев формы:





![{displaystyle pi _ {j} (heta) = pi (heta) ^ {1 / [1 + lambda (j-1)]}, lambda> 0,}](https://wikimedia.org/api/rest_v1/media/math/render/svg/2e9d8c159f93b004aa99ba2ce2249578daf6db1e)
так что первая цепочка - это холодовая цепь с правильной целевой плотностью, а цепочки нагреваются цепи. Обратите внимание, что повышение плотности к власти с имеет эффект выравнивания распределения, как при нагревании металла. В таком распределении легче переходить между пиками (разделенными впадинами), чем в исходном распределении. После каждой итерации предлагается обмен состояниями между двумя случайно выбранными цепочками посредством шага типа Метрополиса. Позволять быть текущим состоянием в цепочке , . Обмен между состояниями цепочек и принимается с вероятностью:








В конце цикла используется продукция только холодовой цепи, а продукция горячей цепи отбрасывается. Эвристически горячие цепи довольно легко посещают локальные пики, а переключение состояний между цепями позволит иногда холодной цепочке перепрыгивать впадины, что приводит к лучшему перемешиванию. Однако если нестабильно, предлагаемые свопы будут приниматься редко. Это причина использования нескольких цепочек, которые отличаются только инкрементально.

Очевидным недостатком алгоритма является то, что цепочки запускаются, и только одна цепочка используется для вывода. По этой причине, идеально подходит для реализации на параллельных машинах, поскольку каждая цепочка обычно требует одинакового количества вычислений на итерацию.


ЛОКАЛЬНЫЙ алгоритм Ларже и Саймона
ЛОКАЛЬНЫЕ алгоритмы[7] предлагает вычислительное преимущество перед предыдущими методами и демонстрирует, что байесовский подход может оценивать неопределенность с практической точки зрения на больших деревьях. ЛОКАЛЬНЫЙ алгоритм является усовершенствованием ГЛОБАЛЬНОГО алгоритма, представленного в Mau, Newton and Larget (1999).[8] в котором все длины ветвей меняются в каждом цикле. ЛОКАЛЬНЫЕ алгоритмы изменяют дерево путем случайного выбора внутренней ветви дерева. Каждый узел на концах этой ветви связан с двумя другими ветвями. По одному из каждой пары выбирается случайным образом. Представьте, что вы берете эти три выбранных края и натягиваете их как веревку для белья слева направо, причем направление (влево / вправо) также выбирается случайным образом. Две конечные точки первой выбранной ветви будут иметь поддерево, висящее как кусок одежды, привязанный к линии. Алгоритм выполняется путем умножения трех выбранных ветвей на обычную случайную величину, что похоже на растяжение или сжатие бельевой веревки. Наконец, крайнее левое из двух подвешенных поддеревьев отсоединяется и снова прикрепляется к веревке для белья в месте, выбранном равномерно случайным образом. Это будет дерево кандидатов.
Предположим, мы начали с выбора внутренней ветви длиной который разделяет таксоны и от остальных. Предположим также, что у нас есть (случайным образом) выбранные ветви длиной и с каждой стороны, и что мы ориентировали эти ветви. Позволять , - текущая длина бельевой веревки. Выбираем новую длину, которая будет , куда равномерная случайная величина на . Тогда для ЛОКАЛЬНОГО алгоритма вероятность принятия может быть вычислена как:










Оценка конвергенции
Чтобы оценить длину ветки 2-таксонового дерева под JC, в котором сайты неизменны и переменны, предположим экспоненциальное априорное распределение со скоростью . Плотность . Вероятности возможных шаблонов сайтов:






для неизменяемых сайтов и

Таким образом, ненормализованное апостериорное распределение:

или, поочередно,

Обновите длину ветви, выбирая новое значение равномерно случайным образом из окна половинной ширины с центром на текущем значении:


куда равномерно распределяется между и . Вероятность приемки:



Пример: , . Сравним результаты для двух значений , и . В каждом случае мы начнем с начальной длины и обновите длину раз.






Максимальная экономия и максимальная вероятность

Существует множество подходов к реконструкции филогенетических деревьев, каждый из которых имеет свои преимущества и недостатки, и однозначного ответа на вопрос «какой метод лучше всего?» Не существует. Максимальная экономия (MP) и максимальная вероятность (ML) - традиционные методы, широко используемые для оценки филогении, и оба используют символьную информацию напрямую, как это делают байесовские методы.
Maximum Parsimony восстанавливает одно или несколько оптимальных деревьев на основе матрицы дискретных символов для определенной группы таксоны и это не требует модели эволюционного изменения. MP дает наиболее простое объяснение заданному набору данных, реконструируя филогенетическое дерево, которое включает в себя как можно меньше изменений в последовательностях; это то, которое демонстрирует наименьшее количество эволюционных шагов для объяснения взаимосвязи между таксонами. Опора ветвей дерева представлена бутстрап процент. По той же причине, по которой он широко использовался, из-за своей простоты, MP также подвергся критике и был отодвинут на задний план ML и байесовскими методами. MP имеет несколько проблем и ограничений. Как показал Фельзенштейн (1978), МП может быть статистически несовместимым,[9] Это означает, что по мере накопления все большего и большего количества данных (например, длины последовательности) результаты могут сходиться в неправильном дереве и приводить к аттракцион длинной ветви, филогенетический феномен, при котором таксоны с длинными ветвями (многочисленные изменения состояния признаков) имеют тенденцию казаться более близкими в филогенезе, чем они есть на самом деле. Что касается морфологических данных, недавние исследования моделирования показывают, что экономия может быть менее точной, чем деревья, построенные с использованием байесовских подходов,[10] потенциально из-за чрезмерной точности,[11] хотя это оспаривается.[12] Исследования с использованием новых методов моделирования показали, что различия между методами вывода являются результатом используемой стратегии поиска и метода консенсуса, а не используемой оптимизации.[13]
Как и в случае максимальной экономии, с максимальной вероятностью будут оцениваться альтернативные деревья. Однако он учитывает вероятность того, что каждое дерево объясняет данные на основе модели эволюции. В этом случае дерево с наибольшей вероятностью объяснения данных выбирается среди других.[14] Другими словами, он сравнивает, как разные деревья предсказывают наблюдаемые данные. Введение модели эволюции в анализ ML дает преимущество перед MP, поскольку учитываются вероятность нуклеотидных замен и скорость этих замен, что более реалистично объясняет филогенетические отношения таксонов. Важным аспектом этого метода является длина ветвей, которую экономия игнорирует, поскольку изменения более вероятны на длинных ветвях, чем на коротких. Такой подход может устранить притяжение длинных ветвей и объяснить большую согласованность ML по сравнению с MP. Хотя многие считают ML лучшим подходом к выводу филогении с теоретической точки зрения, ML требует больших вычислительных ресурсов, и практически невозможно исследовать все деревья, поскольку их слишком много. Байесовский вывод также включает в себя модель эволюции, и основные преимущества перед MP и ML заключаются в том, что он более эффективен в вычислительном отношении, чем традиционные методы, позволяет количественно определять и устранять источник неопределенности и может включать сложные модели эволюции.
Ловушки и разногласия
- Значения начальной загрузки и апостериорные вероятности. Было замечено, что значения поддержки бутстрапа, рассчитанные с учетом экономии или максимального правдоподобия, имеют тенденцию быть ниже, чем апостериорные вероятности, полученные с помощью байесовского вывода.[15][16][17][18] Этот факт приводит к ряду вопросов, таких как: приводят ли апостериорные вероятности к чрезмерной уверенности в результатах? Являются ли значения начальной загрузки более надежными, чем апостериорные вероятности?
- Противоречие использования априорных вероятностей. Использование априорных вероятностей для байесовского анализа многими рассматривается как преимущество, так как дает гипотезе более реалистичное представление о реальном мире. Однако некоторые биологи спорят о субъективности байесовских апостериорных вероятностей после включения этих априорных вероятностей.
- Выбор модели. Результаты байесовского анализа филогении напрямую коррелируют с выбранной моделью эволюции, поэтому важно выбрать модель, которая соответствует наблюдаемым данным, в противном случае выводы филогении будут ошибочными. Многие ученые подняли вопросы об интерпретации байесовского вывода, когда модель неизвестна или неверна. Например, чрезмерно упрощенная модель может дать более высокие апостериорные вероятности.[15][19]
Программное обеспечение MRBAYES
MrBayes - это бесплатный программный инструмент, который выполняет байесовский вывод филогении. Первоначально написано Джоном П. Хуэльзенбеком и Фредериком Ронквистом в 2001 году.[20] По мере роста популярности байесовских методов MrBayes стал одним из предпочтительных программ для многих молекулярных филогенетиков. Он предлагается для операционных систем Macintosh, Windows и UNIX и имеет интерфейс командной строки. Программа использует стандартный алгоритм MCMC, а также вариант MCMC, связанный с Metropolis. MrBayes считывает выровненные матрицы последовательностей (ДНК или аминокислот) в стандарте Формат NEXUS.[21]
MrBayes использует MCMC для аппроксимации апостериорных вероятностей деревьев.[3] Пользователь может изменить предположения модели замещения, априорные значения и детали анализа MC³. Он также позволяет пользователю удалять и добавлять в анализ таксоны и символы. Программа использует самую стандартную модель замены ДНК, 4x4, также называемую JC69, которая предполагает, что изменения по нуклеотидам происходят с равной вероятностью.[22] Он также реализует ряд моделей 20x20 замен аминокислот и кодоновых моделей замены ДНК. Он предлагает различные методы для ослабления предположения о равных скоростях замен по нуклеотидным сайтам.[23] MrBayes также может делать выводы о наследственных состояниях с учетом неопределенности филогенетического дерева и параметров модели.
MrBayes 3 [24] была полностью реорганизованной и реструктурированной версией оригинального MrBayes. Основным нововведением была способность программного обеспечения учитывать неоднородность наборов данных. Эта новая структура позволяет пользователю смешивать модели и использовать преимущества эффективности байесовского анализа MCMC при работе с данными различного типа (например, белками, нуклеотидами и морфологическими данными). По умолчанию он использует Metropolis-Coupling MCMC.
MrBayes 3.2 новая версия MrBayes была выпущена в 2012 году[25] Новая версия позволяет пользователям выполнять несколько анализов параллельно. Он также обеспечивает более быстрые вычисления вероятности и позволяет делегировать эти вычисления блокам обработки графики (GPU). Версия 3.2 предоставляет более широкие возможности вывода, совместимые с FigTree и другими программами просмотра деревьев.
Список программ филогенетики
В эту таблицу включены некоторые из наиболее распространенных филогенетических программ, используемых для вывода филогении в рамках байесовской системы. Некоторые из них не используют исключительно байесовские методы.
| Имя | Описание | Метод | Автор | Ссылка на сайт |
|---|---|---|---|---|
| Платформа рабочего процесса Armadillo | Платформа рабочего процесса, посвященная филогенетическому и общему биоинформатическому анализу | Вывод филогенетических деревьев с использованием метода расстояния, максимального правдоподобия, максимальной экономии, байесовских методов и связанных рабочих процессов | Э. Лорд, М. Леклерк, А. Бок, А.Б. Диалло и В. Макаренков[26] | https://web.archive.org/web/20161024081942/http://www.bioinfo.uqam.ca/armadillo/. |
| Бали-Пхи | Одновременный байесовский вывод о выравнивании и филогении | Байесовский вывод, выравнивание, а также поиск по дереву | Сушард М.А., Ределингс Б.Д.[27] | http://www.bali-phy.org |
| Купание | Байесовский анализ деревьев с генерацией внутренних узлов | Байесовский вывод, демографическая история, разделение населения | И. Дж. Уилсон, Д. Уил, Д. Болдинг [28] | http://www.maths.abdn.ac.uk/˜ijw[постоянная мертвая ссылка ] |
| Байесовская Филогения | Байесовский вывод деревьев с использованием методов Монте-Карло цепи Маркова | Байесовский вывод, множественные модели, смешанная модель (автоматическое разбиение) | М. Пагель, А. Мид[29] | http://www.evolution.rdg.ac.uk/BayesPhy.html |
| PhyloBayes / PhyloBayes MPI | Пробоотборник байесовской цепи Маркова Монте-Карло (MCMC) для филогенетической реконструкции. | Непараметрические методы моделирования межсайтовых вариаций нуклеотидной или аминокислотной склонности. | Н. Лартилло, Н. Родриг, Д. Стаббс, Дж. Ричер [30] | http://www.atgc-montpellier.fr/phylobayes/ |
| ЗВЕРЬ | Деревья выборки байесовского эволюционного анализа | Байесовский вывод, расслабленные молекулярные часы, демографическая история | А. Дж. Драммонд, А. Рамбо и М. А. Сушард [31] | https://beast.community |
| ЗВЕРЬ 2 | Программная платформа для байесовского эволюционного анализа | Байесовский вывод, пакеты, несколько моделей | R Bouckaert, J Heled, D Kühnert, T. Vaughan, CH Wu, D Xie, MA Suchard, A Rambaut, AJ Drummond.[32] | http://www.beast2.org |
| БАКИ | Байесовское соответствие генных деревьев | Байесовское согласование с использованием модифицированного жадного консенсуса некорневых квартетов | К. Ане, Б. Ларже, Д.А. Баум, С. Смит, А. Рокас, Б. Ларгет, С.К. Kotha, C.N. Дьюи, К. Ане [33] | http://www.stat.wisc.edu/~ane/bucky/ |
| Geneious (плагин MrBayes) | Geneious предоставляет инструменты для исследования генома и протеома | Присоединение к соседям, UPGMA, плагин MrBayes, плагин PHYML, плагин RAxML, плагин FastTree, плагин GARLi, плагин PAUP * | A. J. Drummond, M.Suchard, V.Lefort et al. | http://www.geneious.com |
| MrBayes | Филогенетический вывод | Программа для байесовского вывода и выбора модели для широкого спектра филогенетических и эволюционных моделей. | Занг, Хюльзенбек, Дер Марк, Ронквист и Тесленко | https://nbisweden.github.io/MrBayes/ |
| TOPALi | Филогенетический вывод | Выбор филогенетической модели, байесовский анализ и оценка филогенетического дерева максимального правдоподобия, обнаружение сайтов с положительным отбором и анализ местоположения контрольной точки рекомбинации | И.Мильне, Д.Линднер и др.[34] | http://www.topali.org |
Приложения
Байесовский вывод широко используется молекулярными филогенетиками для широкого круга приложений. Некоторые из них включают:

- Вывод филогенеза.[35][36]
- Вывод и оценка неопределенности филогенеза.[37]
- Вывод об эволюции состояния предков.[38][39]
- Вывод ареалов предков.[40]
- Анализ молекулярного датирования.[41][42]
- Модельная динамика диверсификации и исчезновения видов[43]
- Выяснить закономерности распространения патогенов.[44]
Рекомендации
- ^ Лаплас П. (1774 г.). "Memoire sur la Probabilite des Causes par les Evenements". L'Académie Royale des Sciences. 6: 621–656. Английский перевод Стиглер С.М. (1986). «Воспоминание о вероятности причин событий». Статистическая наука. 1 (3): 359–378. Дои:10.1214 / сс / 1177013620.
- ^ Гастингс В.К. (апрель 1970 г.). «Методы выборки Монте-Карло с использованием цепей Маркова и их приложения». Биометрика. 57 (1): 97–109. Bibcode:1970Бимка..57 ... 97Н. Дои:10.1093 / biomet / 57.1.97.
- ^ а б Метрополис N, Розенблют А.В., Розенблют М.Н., Теллер А.Х., Теллер Э. (июнь 1953 г.). «Уравнение состояний вычислений на быстрых вычислительных машинах». Журнал химической физики. 21 (6): 1087–92. Bibcode:1953ЖЧФ..21.1087М. Дои:10.1063/1.1699114.
- ^ Фельзенштейн Дж (2004). Вывод филогении. Сандерленд, Массачусетс: Sinauer Associates.
- ^ Ян З., Раннала Б. (июль 1997 г.). "Байесовский филогенетический вывод с использованием последовательностей ДНК: метод Монте-Карло цепи Маркова". Молекулярная биология и эволюция. 14 (7): 717–24. Дои:10.1093 / oxfordjournals.molbev.a025811. PMID 9214744.
- ^ Гейер CJ (1991). «Марковская цепь Монте-Карло максимальной вероятности». В Keramidas EM, Kaufman SM (ред.). Вычислительная техника и статистика: материалы 23-го симпозиума по интерфейсу. Станция Фэрфакс: Интерфейсный фонд. С. 156–163. OCLC 26603816.
- ^ Ларгет Б., Саймон Д.Л. (июнь 1999 г.). «Алгоритмы Монте-Карло цепи Маркова для байесовского анализа филогенетических деревьев». Молекулярная биология и эволюция. 16 (6): 750–9. Дои:10.1093 / oxfordjournals.molbev.a026160.
- ^ Мау Б., Ньютон М.А., Ларгет Б. (март 1999 г.). «Байесовский филогенетический вывод с помощью методов Монте-Карло цепи Маркова». Биометрия. 55 (1): 1–12. Дои:10.1111 / j.0006-341x.1999.00001.x. PMID 11318142.
- ^ Фельзенштейн Дж (декабрь 1978 г.). «Случаи, в которых методы экономии или совместимости заведомо вводят в заблуждение». Систематическая зоология. 27 (4): 401–10. Дои:10.1093 / sysbio / 27.4.401.
- ^ Castorani MC, Reed DC, Raimondi PT, Alberto F, Bell TW, Cavanaugh KC и др. (Январь 2017 г.). «Колебания плодовитости населения приводят к колебаниям в демографической взаимосвязанности и динамике метапопуляции». Ход работы. Биологические науки. 284 (1847): 20162086. Дои:10.1098 / rspb.2016.2086. ЧВК 5310032. PMID 28123088.
- ^ О'Рейли Дж. Э., Путтик М. Н., Парри Л., Таннер А. Р., Тарвер Дж. Э., Флеминг Дж., Пизани Д., Донохью, ПК (апрель 2016 г.). «Байесовские методы превосходят экономичность, но за счет точности оценки филогении по дискретным морфологическим данным». Письма о биологии. 12 (4): 20160081. Дои:10.1098 / рсбл.2016.0081. ЧВК 4881353. PMID 27095266.
- ^ Голобов П.А., Торрес А., Ариас Дж. С. (2018). «Взвешенная экономия превосходит другие методы филогенетического вывода в моделях, подходящих для морфологии». Кладистика. 34 (4): 407–437. Дои:10.1111 / cla.12205. ISSN 0748-3007.
- ^ Китинг Дж. Н., Сансом Р. С., Саттон М. Д., Найт К. Г., Гарвуд Р. Дж. (Февраль 2020 г.). «Морфологическая филогенетика оценена с использованием новых эволюционных моделей». Систематическая биология. 69 (5): 897–912. Дои:10.1093 / sysbio / syaa012. ЧВК 7440746. PMID 32073641.
- ^ Swofford DL, Olsen GJ, Waddell PJ, Hillis DM (1996). «Филогенетический вывод». В Hillis DM, Moritz C, Mable BK (ред.). Молекулярная систематика, 2-е издание. Сандерленд, Массачусетс: Синауэр. С. 407–514. ISBN 9780878932825.
- ^ а б Сузуки Ю., Глазко Г.В., Ней М. (декабрь 2002 г.). «Чрезмерная достоверность молекулярных филогенезов, полученных байесовской филогенетикой». Труды Национальной академии наук Соединенных Штатов Америки. 99 (25): 16138–43. Bibcode:2002PNAS ... 9916138S. Дои:10.1073 / pnas.212646199. ЧВК 138578. PMID 12451182.
- ^ Альфаро М.Э., Золлер С., Лутцони Ф. (февраль 2003 г.). «Байес или бутстрап? Имитационное исследование, сравнивающее эффективность выборки методом Монте-Карло байесовской цепи Маркова и бутстрапинга при оценке филогенетической достоверности». Молекулярная биология и эволюция. 20 (2): 255–66. Дои:10.1093 / molbev / msg028. PMID 12598693.
- ^ Дуади CJ, Delsuc F, Boucher Y, Doolittle WF, Douzery EJ (февраль 2003 г.). «Сравнение байесовских и бутстрэп-методов максимального правдоподобия филогенетической надежности». Молекулярная биология и эволюция. 20 (2): 248–54. Дои:10.1093 / molbev / msg042. PMID 12598692.
- ^ Гарсия-Сандовал Р. (январь 2014 г.). «Почему некоторые клады имеют низкие частоты начальной загрузки и высокие байесовские апостериорные вероятности». Израильский журнал экологии и эволюции. 60 (1): 41–4. Дои:10.1080/15659801.2014.937900.
- ^ Эриксон П., Свеннблад Б., Бриттон Т., Оксельман Б. (октябрь 2003 г.). «Надежность байесовских апостериорных вероятностей и бутстрэп-частот в филогенетике». Систематическая биология. 52 (5): 665–73. Дои:10.1080/10635150390235485. PMID 14530133.
- ^ Huelsenbeck JP, Ronquist F (август 2001 г.). "MRBAYES: Байесовский вывод филогенетических деревьев". Биоинформатика. Оксфорд, Англия. 17 (8): 754–5. Дои:10.1093 / биоинформатика / 17.8.754. PMID 11524383.
- ^ Мэддисон Д.Р., Суоффорд Д.Л., Мэддисон В.П. (декабрь 1997 г.). «NEXUS: расширяемый формат файлов для систематической информации». Систематическая биология. 46 (4): 590–621. Дои:10.1093 / sysbio / 46.4.590. PMID 11975335.
- ^ Jukes TH, Cantor CR (1969). Эволюция белковых молекул. Нью-Йорк: Academic Press. С. 21–132.
- ^ Ян З (ноябрь 1993 г.). «Оценка максимального правдоподобия филогении по последовательностям ДНК, когда скорости замены различаются по сайтам». Молекулярная биология и эволюция. 10 (6): 1396–401. Дои:10.1093 / oxfordjournals.molbev.a040082. PMID 8277861.
- ^ Ronquist F, Huelsenbeck JP (август 2003 г.). «MrBayes 3: байесовский филогенетический вывод в смешанных моделях». Биоинформатика. Оксфорд, Англия. 19 (12): 1572–4. Дои:10.1093 / биоинформатика / btg180. PMID 12912839.
- ^ Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP (май 2012 г.). «MrBayes 3.2: эффективный байесовский филогенетический вывод и выбор модели в большом модельном пространстве». Систематическая биология. 61 (3): 539–42. Дои:10.1093 / sysbio / sys029. ЧВК 3329765. PMID 22357727.
- ^ Лорд Э, Леклерк М., Бок А, Диалло А.Б., Макаренков В. (2012). «Armadillo 1.1: оригинальная платформа для проектирования и проведения филогенетического анализа и моделирования». PLOS ONE. 7 (1): e29903. Bibcode:2012PLoSO ... 729903L. Дои:10.1371 / journal.pone.0029903. ЧВК 3256230. PMID 22253821.
- ^ Suchard MA, Redelings BD (август 2006 г.). "BAli-Phy: одновременный байесовский вывод о выравнивании и филогении". Биоинформатика (Оксфорд, Англия). 22 (16): 2047–8. Дои:10.1093 / биоинформатика / btl175. PMID 16679334.
- ^ Уилсон И.Дж., Уил М.Э., Лысый ди-джей (июнь 2003 г.). «Выводы из данных ДНК: истории популяции, эволюционные процессы и вероятности совпадения». Журнал Королевского статистического общества: серия A (Статистика в обществе). 166 (2): 155–88. Дои:10.1111 / 1467-985X.00264.
- ^ Пагель М., Мид А. (июнь 2006 г.). "Байесовский анализ коррелированной эволюции дискретных характеров методом обратимой скачкообразной цепи Маркова Монте-Карло". Американский натуралист. 167 (6): 808–25. Дои:10.1086/503444. PMID 16685633. S2CID 205984494.
- ^ Лартильо Н., Филипп Х (июнь 2004 г.). «Модель байесовской смеси для межсайтовых неоднородностей в процессе замены аминокислот». Молекулярная биология и эволюция. 21 (6): 1095–109. Дои:10.1093 / молбев / мш112. PMID 15014145.
- ^ Драммонд AJ, Suchard MA, Xie D, Rambaut A (август 2012 г.). "Байесовская филогенетика с BEAUti and the BEAST 1.7". Молекулярная биология и эволюция. 29 (8): 1969–73. Дои:10.1093 / molbev / mss075. ЧВК 3408070. PMID 22367748.
- ^ Bouckaert R, Heled J, Kühnert D, Vaughan T, Wu CH, Xie D, Suchard MA, Rambaut A, Drummond AJ (апрель 2014 г.). «BEAST 2: программная платформа для байесовского эволюционного анализа». PLOS вычислительная биология. 10 (4): e1003537. Bibcode:2014PLSCB..10E3537B. Дои:10.1371 / journal.pcbi.1003537. ЧВК 3985171. PMID 24722319.
- ^ Ане С., Ларже Б., Баум Д.А., Смит С.Д., Рокас А. (февраль 2007 г.). «Байесовская оценка соответствия между генными деревьями». Молекулярная биология и эволюция. 24 (2): 412–26. Дои:10.1093 / molbev / msl170. PMID 17095535.
- ^ Милн I, Линднер Д., Байер М., Хусмайер Д., Макгуайр Дж., Маршалл Д.Ф., Райт Ф. (январь 2009 г.). «TOPALi v2: богатый графический интерфейс для эволюционного анализа множественных согласований на кластерах HPC и многоядерных рабочих столах». Биоинформатика (Оксфорд, Англия). 25 (1): 126–7. Дои:10.1093 / биоинформатика / btn575. ЧВК 2638937. PMID 18984599.
- ^ Алонсо Р., Кроуфорд А.Дж., Бермингем Е. (март 2012 г.). «Молекулярная филогения эндемичного излучения кубинских жаб (Bufonidae: Peltophryne) на основе митохондриальных и ядерных генов». Журнал биогеографии. 39 (3): 434–51. Дои:10.1111 / j.1365-2699.2011.02594.x.
- ^ Антонелли А., Санмартин I (октябрь 2011 г.). «Массовое вымирание, постепенное охлаждение или быстрое излучение? Реконструкция пространственно-временной эволюции древнего рода покрытосеменных Hedyosmum (Chloranthaceae) с использованием эмпирических и смоделированных подходов». Систематическая биология. 60 (5): 596–615. Дои:10.1093 / sysbio / syr062. PMID 21856636.
- ^ de Villemereuil P, Wells JA, Edwards RD, Blomberg SP (июнь 2012 г.). «Байесовские модели для сравнительного анализа с учетом филогенетической неопределенности». BMC Эволюционная биология. 12: 102. Дои:10.1186/1471-2148-12-102. ЧВК 3582467. PMID 22741602.
- ^ Ронквист Ф (сентябрь 2004 г.). «Байесовский вывод эволюции характера». Тенденции в экологии и эволюции. 19 (9): 475–81. Дои:10.1016 / j.tree.2004.07.002. PMID 16701310.
- ^ Schäffer S, Koblmüller S, Pfingstl T, Sturmbauer C, Krisper G (август 2010 г.). «Реконструкция состояния предков выявляет множественную независимую эволюцию диагностических морфологических признаков в« Высшей Орибатиде »(Акари), противоречащей существующим схемам классификации». BMC Эволюционная биология. 10: 246. Дои:10.1186/1471-2148-10-246. ЧВК 2930640. PMID 20701742.
- ^ Филипович Н., Реннер С.С. (июль 2012 г.). «Brunfelsia (Solanaceae): род, равномерно разделенный между Южной Америкой и радиациями на Кубе и других Антильских островах». Молекулярная филогенетика и эволюция. 64 (1): 1–11. Дои:10.1016 / j.ympev.2012.02.026. PMID 22425729.
- ^ Bacon CD, Baker WJ, Simmons MP (май 2012 г.). «Миоценовое рассеяние вызывает островное излучение в пальмовом трибе Trachycarpeae (Arecaceae)». Систематическая биология. 61 (3): 426–42. Дои:10.1093 / sysbio / syr123. PMID 22223444.
- ^ Сяркинен Т., Бос Л., Олмстед Р.Г., Кнапп С. (сентябрь 2013 г.). «Филогенетическая основа для эволюционного изучения пасленовых (Solanaceae): датированное дерево с тысячей кончиков». BMC Эволюционная биология. 13: 214. Дои:10.1186/1471-2148-13-214. ЧВК 3850475. PMID 24283922.
- ^ Сильвестро Д., Шницлер Дж., Лиоу Л.Х., Антонелли А., Саламин Н. (май 2014 г.). «Байесовская оценка видообразования и исчезновения на основе неполных данных о наличии окаменелостей». Систематическая биология. 63 (3): 349–67. Дои:10.1093 / sysbio / syu006. ЧВК 4361715. PMID 24510972.
- ^ Лемей П., Рамбо А., Драммонд А. Дж., Сушард М. А. (сентябрь 2009 г.). «Байесовская филогеография находит свои корни». PLOS вычислительная биология. 5 (9): e1000520. Bibcode:2009PLSCB ... 5E0520L. Дои:10.1371 / journal.pcbi.1000520. ЧВК 2740835. PMID 19779555.
внешняя ссылка
| Соответствующие поля | ||
|---|---|---|
| Базовые концепты | ||
| Методы вывода | ||
| Текущие темы | ||
| Групповые черты | ||
| Типы групп | ||
| Номенклатура | ||
| ||