Синдром Оменна - Omenn syndrome
| Синдром Оменна | |
|---|---|
 | |
| Синдром Оменна имеет аутосомно-рецессивный тип наследования. | |
| Специальность | Гематология |
Синдром Оменна является аутосомно-рецессивный тяжелый комбинированный иммунодефицит.[1] Это связано с гипоморфный миссенс-мутации в иммунологически значимых генах Т-клетки (и В-клетки ) Такие как гены, активирующие рекомбинацию (RAG1 и RAG2), Рецептор интерлейкина-7-α (IL7Rα), DCLRE1C-Artemis, РМРП-ЧХ, ДНК-лигаза IV, общая гамма-цепочка, WHN-FOXN1, ЗАП-70 и завершить Синдром ДиДжорджи. Без лечения это смертельно.
Симптомы
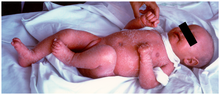
Симптомы очень похожи на болезнь трансплантат против хозяина (РТПХ). Это связано с тем, что у пациентов есть некоторые Т-клетки с ограниченным уровнем рекомбинации с мутантными генами RAG. Эти Т-клетки ненормальны и обладают очень специфическим сродством к аутоантигенам, обнаруженным в тимусе и на периферии. Следовательно, эти Т-клетки являются аутореактивными и вызывают фенотип GVHD.
Характерный симптом - хроническое воспаление кожи, которое проявляется в виде красной сыпи.[2] (раннее начало эритродермия ). Другие симптомы включают: эозинофилия, неспособность процветать, увеличение лимфатических узлов, опухшая селезенка, понос, увеличенная печень, низкий иммуноглобулин уровни (кроме иммуноглобулин E, который повышен), низкий уровень Т-клеток и отсутствие В-клеток.[3]
Генетика
Синдром Оменна вызван частичной потерей функции гена RAG и приводит к симптомам, сходным с синдром тяжелого комбинированного иммунодефицита, включая оппортунистические инфекции. Гены RAG необходимы для рекомбинации генов в Рецептор Т-клеток и B-клеточный рецептор, и потеря этой способности означает, что иммунная система испытывает трудности с распознаванием конкретных патогенов.[2] Синдром Оменна характеризуется потерей функции Т-клеток, что приводит к приживлению материнских лимфоцитов у плода и сосуществованию клонально увеличенных аутологичных и трансплацентарных материнских лимфоцитов.[4]Синдром Оменна иногда может быть вызван другими генами рекомбинации, включая ИЛ-7Rα и RMRP.[3]
Диагностика
Чтобы диагностировать у пациента именно синдром Оменна, аутосомно-рецессивную форму SCID, врач может назначить группу генетического тестирования для поиска микроделеций 22q11 или мутаций генов RAG1 / RAG2.[5]
Уход
Единственное лечение синдрома Оменна - это химиотерапия за которым следует трансплантация костного мозга.[3] Без лечения это быстро приводит к летальному исходу в младенчестве.[2]
Смотрите также
Рекомендации
- ^ Сантагата С., Вилла А, Собакки С., Кортес П., Веццони П. (2000). «Генетические и биохимические основы синдрома Оменна». Иммунол Рев. 178: 64–74. Дои:10.1034 / j.1600-065X.2000.17818.x. PMID 11213808.
- ^ а б c Пархэм, Питер (2009). Иммунная система (3-е изд.). Группа Тейлор и Фрэнсис. п. 128. ISBN 9781136977107.
- ^ а б c Геха, Раиф; Нотаранджело, Луиджи (2012). Тематические исследования в иммунологии: клинический компаньон (6-е изд.). Наука о гирляндах. ISBN 978-0-8153-4441-4.
- ^ Лев А., Саймон А.Дж., Бен-Ари Дж., Такаги Д., Штаубер Т., Трахтенброт Л., Розенталь Е., Рехави Г., Амариглио Н., Сомеч Р. (2014). «Сосуществование клонально размноженных аутологичных и приобретенных трансплацентой материнских Т-клеток в рекомбинации, активирующей тяжелый комбинированный иммунодефицит с генным дефицитом». Клин Эксп Иммунол. 176 (3): 380–6. Дои:10.1111 / cei.12273. ЧВК 4008982. PMID 24666246.
- ^ Министерство здравоохранения и социальных служб США, Информационный центр Национальных институтов здравоохранения, генетических и редких заболеваний (последнее обновление - 2016 г.). Синдром Оменна. Извлекаются из: https://rarediseases.info.nih.gov/diseases/8198/omenn-syndrome
внешняя ссылка
| Классификация | |
|---|---|
| Внешние ресурсы |