Синдром Вискотта – Олдрича - Wiskott–Aldrich syndrome
| Синдром Вискотта-Олдрича | |
|---|---|
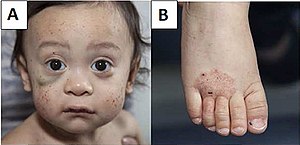 | |
| А) Множественные петехии лица и гематома под правым глазом (слева на снимке). Б) Экзема стопы. | |
| Специальность | Иммунология |
Синдром Вискотта – Олдрича (БЫЛ) является редким Х-сцепленный рецессивный болезнь, характеризующаяся экзема, тромбоцитопения (низкий тромбоцит считать), иммунодефицит и кровавый понос (вторичный по отношению к тромбоцитопении).[1] Его также иногда называют экзема-тромбоцитопения-синдром иммунодефицита в соответствии с первоначальным описанием Олдрича в 1954 году.[2] Связанные с WAS расстройства Х-сцепленная тромбоцитопения (XLT) и Х-связанная врожденная нейтропения (XLN) могут иметь похожие, но менее серьезные симптомы и вызваны мутациями одного и того же гена.
Признаки и симптомы
WAS чаще всего встречается у мужчин из-за его X-сцепленного рецессивного характера наследования, затрагивая от 1 до 10 мужчин на миллион.[1] Первые признаки обычно петехии и синяки в результате низкого количества тромбоцитов (т. е. тромбоцитопения ). Спонтанный кровотечение из носа и кровавый понос также распространены и экзема обычно развивается в течение первого месяца жизни. Рецидивирующий бактериальный инфекции развиваются к трем месяцам. У большинства детей с WAS развивается хотя бы один аутоиммунное заболевание, и раки (в основном лимфома и лейкемия ) развиваются примерно у трети пациентов.[3] Иммуноглобулин М (IgM) уровни снижаются, IgA и IgE подняты, и IgG уровни могут быть нормальными, пониженными или повышенными.[4] Помимо тромбоцитопении, у пациентов с СВО наблюдаются аномально маленькие тромбоциты (т. Е. Микротромбоциты) и ~ 30% также имеют повышенное эозинофил подсчитывает (т.е. эозинофилия ).[5]
Патофизиология
Микротромбоциты, наблюдаемые у пациентов с WAS, наблюдались только при одном другом состоянии, ARPC1B дефицит.[6] В обоих случаях считается, что дефектные тромбоциты удаляются из кровотока посредством селезенка и / или печень, что приводит к снижению количества тромбоцитов. Пациенты с СВО имеют повышенную восприимчивость к инфекциям, особенно ушей и носовых пазух, и этот иммунный дефицит был связан с уменьшением антитело производство и неспособность иммунной Т-клетки для эффективной борьбы с инфекцией.[7]
Генетика

WAS связан с мутациями в ген на короткой руке Х хромосома (Xp11.23), который изначально назывался Белок синдрома Вискотта-Олдрича ген и официально известен как БЫЛ (Идентификатор гена: 7454).[8] Х-сцепленная тромбоцитопения также связана с БЫЛ мутации, хотя они и отличаются от тех, которые вызывают полноценный WAS. Редкое заболевание, связанное с Х-сцепленной нейтропенией, также было связано с определенной подгруппой БЫЛ мутации.[9]
Белковый продукт БЫЛ известен как WASp. Он содержит 502 аминокислоты и в основном выражается в кроветворный клетки (клетки костного мозга, которые развиваются в клетки крови). Основная функция WASp - активировать полимеризацию актина, выступая в качестве фактора, способствующего зародышеобразованию (NPF) для Комплекс Арп2 / 3, который генерирует разветвленные актиновые филаменты. Некоторые белки могут служить NPF, и было замечено, что в тромбоцитах WAS комплекс Arp2 / 3 функционирует нормально, что указывает на то, что WASp не требуется для его активации в тромбоцитах.[10] В Т-клетках WASp важен, потому что, как известно, он активируется через Рецептор Т-клеток сигнальные пути для индукции кортикального актина цитоскелет перестройки, ответственные за формирование иммунологический синапс.[11]
Тяжесть симптомов, вызванных БЫЛ мутации коррелируют с их влиянием на WASp. Аллели, которые не производят или не производят усеченный белок, имеют более серьезные эффекты, чем миссенс-мутации.[12] Хотя аутоиммунные заболевания и злокачественные новообразования могут возникать при обоих типах мутаций, пациенты с усеченными Оса несут более высокий риск. Дефект в CD43 молекула также была обнаружена у пациентов с WAS.[13]
Диагностика
Диагноз ставится на основании клинических показателей, мазок периферической крови, и низкий иммуноглобулин уровни. Обычно уровни IgM низкие, уровни IgA повышены, а уровни IgE могут быть повышены; парапротеины изредка наблюдаются.[14] Иммунологическое обследование кожи (тестирование на аллергию) может выявить пониженную чувствительность. Не все пациенты имеют положительный семейный анамнез заболевания; новые мутации действительно происходят. Часто лейкоз можно заподозрить на основании низкого уровня тромбоцитов и инфекций, и биопсия костного мозга может быть выполнено. Обычно наблюдается снижение уровня WASp. Текущий золотой стандарт диагностики - геномный. Анализ последовательности ДНК, который может выявить WAS и связанные с ним расстройства XLT и XLN у подавляющего большинства пациентов и носителей.[нужна цитата ]
Классификация
Jin et al. (2004) используют числовую оценку степени тяжести:[12]
- 0,5: перемежающаяся тромбоцитопения
- 1.0: тромбоцитопения и мелкие тромбоциты (микротромбоцитопения)
- 2.0: микротромбоцитопения плюс нормально чувствительная экзема или случайные инфекции верхних дыхательных путей
- 2.5: микротромбоцитопения плюс поддающаяся лечению, но тяжелая экзема или инфекции дыхательных путей, требующие антибиотиков
- 3.0: микротромбоцитопения плюс экзема и инфекции дыхательных путей, требующие антибиотиков.
- 4.0: микротромбоцитопения плюс экзема, постоянно требующая лечения, и / или тяжелые или опасные для жизни инфекции
- 5.0: микротромбоцитопения плюс аутоиммунное заболевание или злокачественное новообразование
Уход
В настоящее время лечение синдрома Вискотта – Олдрича основано на коррекции симптомов. Аспирин и другие нестероидные противовоспалительные препараты следует избегать, так как они могут нарушить функцию тромбоцитов, которая уже нарушена. Защитный шлем может защитить детей от кровотечение в мозг которые могут возникнуть в результате травм головы. При очень низком уровне тромбоцитов пациентам может потребоваться переливание тромбоцитов или удаление селезенки. Для пациентов с частыми инфекциями внутривенные иммуноглобулины (ВВИГ) можно вводить для усиления иммунной системы. Анемия от кровотечения может потребоваться добавка железа или переливание крови.[нужна цитата ]
Поскольку WAS - это в первую очередь заболевание кроветворных тканей, кроветворный стволовая клетка трансплантация, осуществленная через пуповинная кровь или же пересадка костного мозга предлагает единственную надежду на излечение. Это может быть рекомендовано пациентам с HLA -идентичные доноры, совпадающие братья или сестры-доноры или даже в случаях неполных совпадений, если пациенту 5 лет или меньше.[нужна цитата ]
Исследования коррекции синдрома Вискотта – Олдрича с помощью генная терапия используя лентивирус начались.[15][16]Доказательство принципа успешной генной терапии гемопоэтическими стволовыми клетками было предоставлено пациентам с синдромом Вискотта – Олдрича.[17]В настоящее время многие исследователи продолжают разрабатывать оптимизированные векторы для генной терапии.[12][15][16][18] В июле 2013 года итальянский институт генной терапии San Raffaele Telethon (HSR-TIGET) сообщил, что у троих детей с синдромом Вискотта – Олдрича наблюдалось значительное улучшение через 20–30 месяцев после лечения генетически модифицированным лентивирусом.[19] В апреле 2015 года результаты последующего исследования во Франции и Великобритании, в котором шестеро детей с синдромом Вискотта – Олдрича лечились генной терапией, были признаны многообещающими.[20][21] Среднее время наблюдения составило 27 месяцев.
Прогноз
Исходы синдрома Вискотта-Олдрича сильно различаются в зависимости от того, насколько серьезно поражен человек, при этом легкая часть спектра заболевания связана с БЫЛ ген, называемый Х-сцепленная тромбоцитопения или Х-сцепленная нейтропения. Синдром Вискотта – Олдрича связан с переменная выразительность Это означает, что даже в пределах одной семьи некоторые могут проявлять только хроническую тромбоцитопению, в то время как другие испытывают серьезные, опасные для жизни осложнения синдрома Вискотта-Олдрича в младенчестве или детстве.[22][23] Учитывая, что симптомы часто прогрессируют с возрастом, сложно предсказать, насколько серьезно пострадает ребенок с новым диагнозом. Продолжительность жизни при Х-связанной тромбоцитопении считается нормальной, с сообщениями о мужчинах с минимальным поражением в седьмой десяток лет.[24] Существует некоторая корреляция генотип-фенотип: у большинства людей с Х-связанной тромбоцитопенией неверные варианты в БЫЛ ген по сравнению с 86,5% тех, кто не продуцирует WASp, имеют фенотип синдрома Вискотта-Олдрича.[25] В целом прогноз для людей с синдромом Вискотта – Олдрича значительно улучшился за последние несколько десятилетий благодаря более раннему диагнозу и большему доступу к вариантам лечения, таким как трансплантация гемопоэтических стволовых клеток.
Эпидемиология
По оценкам, заболеваемость синдромом Вискотта – Олдрича в Соединенных Штатах составляет 1 случай на 250 000 живорождений мужского пола. Никакого географического фактора нет.[26]
История
Синдром назван в честь доктора Альфреда Вискотта (1898–1978), немецкого педиатра, который впервые заметил синдром в 1937 году.[27] и доктор Роберт Андерсон Олдрич (1917–1998), американский педиатр, описавший болезнь в семье американцев голландского происхождения в 1954 году.[2] Вискотт описал трех братьев с похожей болезнью, сестры которых не пострадали. В 2006 году немецкая исследовательская группа проанализировала членов семей трех случаев Вискотта и предположила, что они, вероятно, имеют новую мутацию сдвига рамки считывания первого экзона Оса ген.[28]
Рекомендации
- ^ а б Справка, Дом генетики. «Синдром Вискотта-Олдрича». Домашний справочник по генетике. Получено 2016-06-26.
- ^ а б Олдрич Р.А., Стейнберг А.Г., Кэмпбелл, округ Колумбия (февраль 1954 г.). «Родословная, демонстрирующая рецессивное состояние, связанное с полом, характеризующееся истощением ушей, экзематоидным дерматитом и кровавой диареей». Педиатрия. 13 (2): 133–9. PMID 13133561.
- ^ Синдром Вискотта-Олдрича в eMedicine
- ^ Санде М.А., Уилсон В.П. (2001). Текущая диагностика и лечение инфекционных заболеваний. Нью-Йорк: Lange Medical Books / McGraw-Hill. п. 361. ISBN 978-0-8385-1494-8.[страница нужна ]
- ^ Наваби Б., Аптон Дж. Э. (2016). «Первичные иммунодефициты, связанные с эозинофилией». Аллергия, астма и клиническая иммунология. 12: 27. Дои:10.1186 / s13223-016-0130-4. ЧВК 4878059. PMID 27222657.
- ^ Кар У.Х., Плутеро Ф.Г., Элькадри А., Уорнер Н., Дробак М, Чен СН, Ло РВ, Ли Л, Ли Р, Ли Кью, Тхени С., Пан Дж., Леунг Дж., Лара-Корралес I, Мурчи Р., Катц Э, Лаксер Р.М., Аптон Дж., Ройфман К.М., Йунг Р.С., Брюмелл Дж. Х., Муис А.М. (апрель 2017 г.). «Потеря компонента комплекса Arp2 / 3 ARPC1B вызывает аномалии тромбоцитов и предрасполагает к воспалительным заболеваниям». Nature Communications. 8: 14816. Bibcode:2017НатКо ... 814816K. Дои:10.1038 / ncomms14816. ЧВК 5382316. PMID 28368018.
- ^ "Синдром Вискотта-Олдрича: иммунодефицитные расстройства: руководство Merck Professional". Получено 2008-03-01.
- ^ Дерри JM, Ochs HD, Francke U (август 1994). «Выделение нового гена, мутировавшего при синдроме Вискотта-Олдрича». Клетка. 78 (4): 635–44. Дои:10.1016/0092-8674(94)90528-2. PMID 8069912. S2CID 9040376.
- ^ Вестерберг Л.С., Меелу П., Баптиста М., Эстон М.А., Адамович Д.А., Котта-де-Алмейда В., Сид Б., Розен М.К., Ванденберг П., Трэшер А.Дж., Кляйн С., Альт FW, Снаппер С.Б. «Активация мутаций WASP, связанных с Х-сцепленной нейтропенией, приводит к усилению полимеризации актина, измененным ответам цитоскелета и геномной нестабильности в лимфоцитах». Журнал экспериментальной медицины. 207 (6): 1145–52. Дои:10.1084 / jem.20091245. ЧВК 2882832. PMID 20513746.
- ^ Falet H, Hoffmeister KM, Neujahr R, Hartwig JH (сентябрь 2002 г.). «Нормальная активация комплекса Arp2 / 3 в тромбоцитах, лишенных WASp». Кровь. 100 (6): 2113–22. Дои:10.1182 / blood.V100.6.2113. PMID 12200375.
- ^ Малинова Д., Фриче М., Новосад С.Р., Армер Х., Манро П.М., Бланделл М.П., Чаррас Г., Толар П., Баума Г., Трэшер А.Дж. (май 2016 г.). «WASp-зависимая стабильность актинового цитоскелета в иммунологическом синапсе дендритных клеток необходима для обширных функциональных контактов с Т-клетками». Журнал биологии лейкоцитов. 99 (5): 699–710. Дои:10.1189 / jlb.2a0215-050rr. ЧВК 5404712. PMID 26590149.
- ^ а б c Джин И, Мазза С., Кристи Дж. Р., Джилиани С., Фиорини М., Мелла П., Ганделлини Ф., Стюарт Д. М., Чжу К., Нельсон Д. Л., Нотаранджело Л. Д., Охс HD (декабрь 2004 г.). «Мутации белка синдрома Вискотта-Олдрича (WASP): горячие точки, влияние на транскрипцию, трансляцию и корреляцию фенотипа / генотипа». Кровь. 104 (13): 4010–9. Дои:10.1182 / кровь-2003-05-1592. PMID 15284122.
- ^ Розенштейн Ю., Парк Дж. К., Хан В. К., Розен Ф. С., Бирер Б. Е., Буракофф С. Дж. (Ноябрь 1991 г.). «CD43, молекула, дефектная при синдроме Вискотта-Олдрича, связывает ICAM-1». Природа. 354 (6350): 233–5. Bibcode:1991Натура.354..233R. Дои:10.1038 / 354233a0. PMID 1683685. S2CID 4256329.
- ^ Радл Дж., Дурен Л.Х., Морелл А., Скварил Ф., Фоссен Дж. М., Уиттенбогаарт СН (август 1976 г.). «Иммуноглобулины и временные парапротеины в сыворотке крови пациентов с синдромом Вискотта-Олдрича: последующее исследование». Клиническая и экспериментальная иммунология. 25 (2): 256–63. ЧВК 1541349. PMID 954233.
- ^ а б Гали А., Ронкароло М.Г., Трэшер А.Дж. (февраль 2008 г.). «Разработка лентивирусной генной терапии синдрома Вискотта-Олдрича». Мнение эксперта по биологической терапии. 8 (2): 181–90. Дои:10.1517/14712598.8.2.181. ЧВК 2789278. PMID 18194074.
- ^ а б Frecha C, Toscano MG, Costa C, Saez-Lara MJ, Cosset FL, Verhoeyen E, Martin F (июнь 2008 г.). «Улучшенные лентивирусные векторы для генной терапии синдрома Вискотта-Олдрича имитируют профили эндогенной экспрессии на протяжении всего гематопоэза». Генная терапия. 15 (12): 930–41. Дои:10.1038 / gt.2008.20. PMID 18323794.
- ^ Boztug K, Schmidt M, Schwarzer A, Banerjee PP, Díez IA, Dewey RA, Böhm M, Nowrouzi A, Ball CR, Glimm H, Naundorf S, Kühlcke K, Blasczyk R, Kondratenko I, Maródi L, Orange JS, von Kalle C, Кляйн C (ноябрь 2010 г.). «Генная терапия стволовыми клетками при синдроме Вискотта-Олдрича». Медицинский журнал Новой Англии. 363 (20): 1918–27. Дои:10.1056 / NEJMoa1003548. ЧВК 3064520. PMID 21067383.
- ^ Дьюи Р.А., Аведильо Диес И., Баллмайер М., Филипович А., Грейл Дж., Гюнгёр Т., Хаппель С., Машан А., Ноян Ф., Паннике Ю., Шварц К., Снаппер С., Велте К., Кляйн С. (сентябрь 2006 г.). «Ретровирусный перенос гена WASP в гемопоэтические стволовые клетки человека восстанавливает актиновый цитоскелет в миелоидных потомственных клетках, дифференцированных in vitro». Экспериментальная гематология. 34 (9): 1161–9. Дои:10.1016 / j.exphem.2006.04.021. PMID 16939809.
- ^ Aiuti A, Biasco L, Scaramuzza S, Ferrua F, Cicalese MP, Baricordi C, Dionisio F, Calabria A, Giannelli S, Castiello MC, Bosticardo M, Evangelio C, Assanelli A, Casiraghi M, Di Nunzio S, Callegaro L, Benati C, Риццарди П., Пеллин Д., Ди Серио С., Шмидт М., Фон Калле С., Гарднер Дж., Мехта Н., Недува В., Доу Д. Д., Гали А., Миньеро Р., Финокки А., Метин А., Банерджи П. П., Orange JS, Галимберти S, Valsecchi MG, Biffi A, Montini E, Villa A, Ciceri F, Roncarolo MG, Naldini L (август 2013 г.). «Генная терапия лентивирусными гемопоэтическими стволовыми клетками у пациентов с синдромом Вискотта-Олдрича». Наука. 341 (6148): 1233151. Дои:10.1126 / наука.1233151. ЧВК 4375961. PMID 23845947.
- ^ Галлахер, Джеймс (21 апреля 2015 г.) Генная терапия: «Приручить ВИЧ» для лечения болезней BBC News, Health, по состоянию на 21 апреля 2015 г.
- ^ Malech HL, Ochs HD (апрель 2015 г.). «Наступающая эра клинических преимуществ генной терапии». JAMA. 313 (15): 1522–3. Дои:10.1001 / jama.2015.2055. PMID 25898049.
- ^ Бухбиндер, Дэвид; Надо, Кари; Наджент, Дайан (2011-06-28). «Монозиготная пара близнецов, демонстрирующая дискордантный фенотип Х-связанной тромбоцитопении и синдрома Вискотта – Олдрича: роль для эпигенетики?». Журнал клинической иммунологии. 31 (5): 773–777. Дои:10.1007 / s10875-011-9561-3. ISSN 0271-9142.
- ^ Альберт, Майкл Н; Нотаранджело, Луиджи Д; Охс, Ханс Д. (январь 2011 г.). «Клинический спектр, патофизиология и лечение синдрома Вискотта – Олдрича». Текущее мнение в гематологии. 18 (1): 42–48. Дои:10.1097 / moh.0b013e32834114bc. ISSN 1065-6251.
- ^ Альберт, Майкл Х .; Bittner, Tanja C .; Нонояма, Сигэаки; Нотаранджело, Люсия Дора; Бернс, Шивон; Имаи, Косуке; Espanol, Teresa; Фаст, Андерс; Пелье, Изабель; Штраус, Габриэле; Морио, Томохиро (22 апреля 2010 г.). «Х-сцепленная тромбоцитопения (XLT) из-за мутаций WAS: клинические характеристики, долгосрочные результаты и варианты лечения». Кровь. 115 (16): 3231–3238. Дои:10.1182 / кровь-2009-09-239087. ISSN 0006-4971.
- ^ Имаи, Косуке; Нонояма, Сигэаки; Охс, Ханс Д. (декабрь 2003 г.). «Мутации и фенотип гена WASP (белок синдрома Вискотта-Олдрича)». Текущее мнение в области аллергии и клинической иммунологии. 3 (6): 427–436. Дои:10.1097/00130832-200312000-00003. ISSN 1528-4050.
- ^ Роберт А. Шварц, доктор медицины, магистр здравоохранения; Главный редактор: Харуми Джионучи, доктор медицины. Детский синдром Вискотта-Олдрича
- ^ Вискотт, А (1937). "Familiärer, angeborener Morbus Werlhofii? («Семейная врожденная болезнь Верльгофа?») ». Montsschr Kinderheilkd. 68: 212–16.
- ^ Биндер В., Альберт М.Х., Кабус М., Бертоне М., Майндл А., Белоградский Б.Х. (октябрь 2006 г.). «Генотип исходного фенотипа Вискотта». Медицинский журнал Новой Англии. 355 (17): 1790–3. Дои:10.1056 / NEJMoa062520. PMID 17065640. S2CID 30040802.
дальнейшее чтение
- GeneReviews / NIH / NCBI / UW запись о WAS-связанных заболеваниях, включая синдром Вискотта-Олдрича WAS X-связанная тромбоцитопения XLT и X-связанная врожденная нейтропения XLN
- Фонд иммунодефицита - Глава VII, «Синдром Вискотта-Олдрича»
внешняя ссылка
| Классификация | |
|---|---|
| Внешние ресурсы |