Синдром Леша – Найхана - Lesch–Nyhan syndrome
| Синдром Леша – Найхана | |
|---|---|
| Другие имена | Ювенильная подагра[1] |
| Специальность | Эндокринология |
Синдром Леша – Найхана (LNS) является редким наследственное заболевание вызвано нехваткой фермент гипоксантин-гуанинфосфорибозилтрансфераза (HGPRT). Этот дефицит возникает из-за мутации в HPRT1 ген расположен на Х хромосома. LNS поражает примерно 1 из 380 000 живорождений.[2] Расстройство было впервые обнаружено и клинически охарактеризовано американским студентом-медиком. Майкл Леш и его наставник, педиатр Уильям Найхан, в Джонс Хопкинс.[3]
Дефицит HGPRT вызывает накопление мочевая кислота во всех жидкостях организма. Комбинация повышенного синтеза и пониженного использования пурины приводит к высокому уровню выработки мочевой кислоты. Это приводит как к высокий уровень мочевой кислоты в крови и моча, связанные с тяжелым подагра и проблемы с почками. Неврологические признаки включают плохой мышечный контроль и умеренное Интеллектуальная недееспособность. Эти осложнения обычно появляются на первом году жизни. Начиная со второго года жизни, особенно яркой особенностью LNS является нанесение себе увечий поведение, характеризующееся кусанием губ и пальцев. Неврологические симптомы включают гримасу на лице, непроизвольные корчи и повторяющиеся движения рук и ног, подобные тем, которые наблюдаются при болезнь Хантингтона. В причина неврологических отклонений остается неизвестным. Поскольку недостаток HGPRT приводит к тому, что организм плохо использует витамин B12, у некоторых мужчин может развиться мегалобластная анемия.[4]
LNS наследуется в Х-сцепленный рецессивный манера; мутация гена обычно переносится матерью и передается ее сыну, хотя одна треть всех случаев возникает de novo (от новых мутаций) и не имеют семейного анамнеза. LNS - это присутствует при рождении у мальчиков. Большинство, но не все, люди с этим дефицитом имеют серьезные психические и физические проблемы на протяжении всей жизни. Есть несколько редких случаев в мире пораженных женщин.[5]
Симптомы, вызванные накоплением мочевой кислоты (подагра и почка симптомы) хорошо поддаются лечению такими лекарствами, как аллопуринол снижающие уровень мочевой кислоты в крови. Психические отклонения и членовредительство плохо поддаются лечению. Лечения нет, но многие люди доживают до взрослой жизни. Несколько новых экспериментальных методов лечения могут облегчить симптомы.
Признаки и симптомы
LNS характеризуется тремя основными отличительными чертами: неврологический дисфункция, познавательный и поведенческий беспорядки включая членовредительство, и мочевая кислота перепроизводство (гиперурикемия ). Повреждение базальный ганглий заставляет больных принимать характерную стойку для ограждения из-за характера поражения. Некоторые также могут быть поражены макроцитарная анемия. Практически все пациенты - мужчины; самцы страдают задержкой роста и половое созревание, и у большинства развиваются сморщенные яички или атрофия яичек. Женщины-носители подвергаются повышенному риску подагрический артрит но обычно на них не влияет.[нужна цитата ]
Избыточное производство мочевой кислоты
Одним из первых симптомов заболевания является наличие песчаных кристаллов мочевая кислота в подгузниках пострадавшего младенца. Избыточное производство мочевой кислоты может привести к образованию кристаллов мочевой кислоты или камней в почки, мочеточники, или же мочевой пузырь. Такие кристаллы, откладывающиеся в суставах позже при болезни, могут вызывать подагра -подобно артрит с отеком и болезненностью. Избыточное производство мочевой кислоты присутствует при рождении, но не может быть обнаружено обычными методами клинических лабораторных исследований. Концентрация мочевой кислоты в сыворотке часто бывает нормальной, поскольку избыток пуринов быстро выводится с мочой. Кристаллы обычно выглядят как оранжевый зернистый материал, или они могут сливаться, образуя несколько крошечных камней или отдельные большие камни, которые трудно пройти. Камни или камни обычно вызывают гематурия (кровь в моче) и увеличивают риск инфекция мочевыводящих путей. Некоторые жертвы страдают повреждением почек из-за таких камни в почках. Камни могут быть признаком болезни, но могут оставаться незамеченными в течение месяцев или даже лет.[нужна цитата ]
Нарушение нервной системы
Периоды до и после родов обычно нормальны для людей с LNS. Наиболее частые предлежания аномально уменьшены мышца тон (гипотония ) и задержка в развитии, которая проявляется в возрасте от трех до шести месяцев. Больные поздно садятся, в то время как большинство из них никогда не ползают или ходить. Отсутствие речь также очень распространенная черта, связанная с LNS.[нужна цитата ]
Раздражительность чаще всего отмечается вместе с первыми признаками поражения нервной системы. В течение первых нескольких лет жизни экстрапирамидный вовлечение вызывает ненормальные непроизвольные сокращения мышц, такие как потеря моторного контроля (дистония ), корчащимися движениями (хореоатетоз ) и выгибание позвоночника (опистотонус ). Признаки пирамидальная система вовлечение, в том числе спастичность, гиперактивные рефлексы (гиперрефлексия ) и разгибатель подошвенные рефлексы, тоже случаются. Сходство с атетоидом церебральный паралич проявляется в неврологических аспектах LNS. В результате у большинства людей изначально диагностируется церебральный паралич. Двигательная инвалидность настолько обширна, что большинство людей никогда не ходят и на всю жизнь становятся инвалидами.[нужна цитата ]
Самоповреждающее поведение
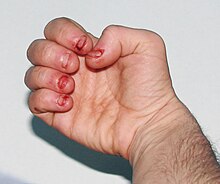
Пострадавшие имеют когнитивные нарушения и поведенческие расстройства, которые проявляются в возрасте от двух до трех лет. Неуправляемый членовредительство связанные с LNS также обычно начинаются в трехлетнем возрасте. Самоповреждение начинается с прикусывания губ и языка; По мере прогрессирования заболевания у больных часто появляются кусания пальцев и тряски головой.[6] Самоповреждение может усиливаться во время стресса. Самоповреждение является отличительной чертой болезни и проявляется у 85% пораженных мужчин.[7]
Большинство людей страдают когнитивными нарушениями, которые иногда трудно отличить от других симптомов из-за поведенческих нарушений и двигательных нарушений, связанных с синдромом. Во многих отношениях такое поведение может рассматриваться как психологическое продолжение принуждения к самоповреждению и включать отказ от желаемых угощений или путешествий, отплату за доброту холодностью или гневом, неспособность правильно ответить на вопросы теста, несмотря на учебу и желание добиться успеха. , провоцируя гнев у опекунов, когда хочется привязанности.[7]
Также встречается компульсивное поведение, в том числе агрессивность, рвота, плевать и копролалия (непроизвольная ругань). Развитие этого типа поведения иногда наблюдается в течение первого года жизни или в раннем детстве, но у других оно может развиться только в более позднем возрасте.
LNS у женщин
В то время как самки-носители обычно бессимптомное состояние, у них действительно увеличивается выведение мочевой кислоты, а у некоторых могут развиться симптомы гиперурикемия и в последние годы страдают подагрой. В этом контексте тестирование не имеет клинических последствий, но может выявить возможность передачи признака детям мужского пола. Женщинам также может потребоваться тестирование, если у ребенка мужского пола развивается LNS. В этом случае отрицательный результат теста означает, что болезнь сына является результатом новой мутации, и риск у братьев и сестер не увеличивается.[нужна цитата ]
Женщины, несущие одну копию дефектного гена, являются носителями с 50% вероятностью передачи болезни своим сыновьям. Чтобы затронуть самку, ей необходимо иметь две копии мутировавшего гена, одна из которых унаследована от ее отца. Мужчины, страдающие LNS, обычно не имеют детей из-за изнуряющих последствий болезни. Самка может унаследовать Х-хромосому от своего здорового отца, который несет новую мутацию гена HGPRT. При таких обстоятельствах девочка может родиться с LNS, и хотя есть несколько сообщений об этом, это происходит очень редко. Подавляющее большинство пациентов с LNS - мужчины.[нужна цитата ]
Менее тяжелые формы
Менее тяжелое родственное заболевание, частичный дефицит HPRT, известно как синдром Келли – Сигмиллера (синдром Леша – Найхана включает полный дефицит HPRT). Симптомы обычно связаны с меньшим неврологическим поражением, но болезнь по-прежнему вызывает подагру и камни в почках.[8]
Генетика

LNS из-за мутации в HPRT1 ген,[2][9] назван так потому, что кодирует фермент гипоксантин-гуанинфосфорибозилтрансфераза (HPRT или HGPRT, EC 2.4.2.8 ). Этот фермент участвует в биохимических процессах, которые организм использует для производства пурины, один из компонентов ДНК и РНК. Дефекты этого фермента приводят к увеличению производства мочевая кислота. Поскольку HPRT ген находится на Х хромосома, LNS - это Х-связанный наследственное заболевание.[нужна цитата ]
Отец заболевшего самца не будет носителем мутанта. аллель, и не будет болезни. An обязательный перевозчик будет женщина, у которой есть больной сын и еще один больной родственник по материнской линии.
Если женщина первая в семье имеет больного сына, Правило холдейна предсказывает 2/3 шанса, что она является носителем, и 1/3 шанса, что у сына будет новый мутация зародышевой линии.[нужна цитата ]
Риск для братьев и сестер пострадавшего человека зависит от статуса носителя самой матери. Любая женщина-носитель имеет 50% шанс передать мутацию HPRT1 в каждом беременность. Сыновья, унаследовавшие мутацию, будут затронуты, в то время как дочери, унаследовавшие мутацию, будут носителями. Следовательно, с каждой беременностью у женщины-носителя есть 25% -ный шанс иметь затронутого мужчину, 25% -ный шанс иметь женщину-носителя и 50% -ный шанс иметь нормального мужчину или женщину.[нужна цитата ]
Самцы с LNS не размножаются из-за особенностей заболевания. Однако, если мужчина с менее тяжелым фенотип воспроизводит, все его дочери являются носителями, и ни один из его сыновей не пострадает.[нужна цитата ]
Патофизиология

Как и в других Х-связанный болезни, мужчины страдают, потому что у них есть только одна копия Х-хромосомы. При синдроме Леша-Найхана дефектным геном является ген гипоксантин-гуанинфосфорибозилтрансфераза (HPRT), участник «утилизации» пурин нуклеотиды. Женщины-носители имеют вторую Х-хромосому, которая содержит «нормальную» копию HPRT, предотвращающую развитие болезни, хотя у них может быть повышенный риск гиперурикемии.
Известно большое количество мутаций HPRT. Мутации, которые лишь незначительно снижают функцию фермента, обычно не вызывают тяжелую форму LNS, но действительно вызывают более легкую форму заболевания, которая все еще характеризуется избыточной продукцией пуринов, сопровождаемой восприимчивостью подагра и мочевая кислота нефролитиаз.
Формирование ДНК (во время деление клеток ) требует нуклеотиды, молекулы, которые являются строительными блоками для ДНК. Пуриновые основания (аденин и гуанин ) и пиримидин базы (тимин и цитозин ) связаны с дезоксирибозой и фосфатом и при необходимости включаются. Обычно нуклеотиды синтезируются de novo из аминокислоты и другие прекурсоры. Однако небольшая часть «перерабатывается» из деградированной ДНК разрушенных клеток. Это называется «спасательный путь».
HGPRT - это «фермент спасения» пуринов: он направляет гипоксантин и гуанин обратно в синтез ДНК. Отказ этого фермента приводит к двум результатам:
- Продукты распада клеток не могут быть повторно использованы, и поэтому они разлагаются. Это приводит к увеличению мочевая кислота, продукт распада пурина.
- В de novo путь стимулируется из-за избытка PRPP (5-фосфо-D-рибозил-1-пирофосфат или просто фосфорибозилпирофосфат).
Ранее было неясно, были ли неврологические нарушения в LNS связаны с нейротоксичностью мочевой кислоты или с относительной нехваткой «нового» пурина. нуклеотиды во время основных этапов синтеза. Генетические мутации, влияющие на ферменты de novo Путь синтеза, возможно, может способствовать заболеванию, хотя они редки или неизвестны. Мочевая кислота была предложена как возможная причина нейротоксичности, но это не доказано.
Важно отметить, что данные свидетельствуют о том, что одно или несколько поражений в полосатый дофаминергический проводящие пути могут иметь центральное значение для неврологического дефицита, особенно хореоатетоидной дискинезии и членовредительства.[10][11][12] 6-гидроксидофамин токсичность для грызунов может быть полезной животной моделью для синдрома, хотя это не доказано.[13]Однако связь между синтезом дофамина и пурина - это нуклеотид, называемый гуанозинтрифосфат или «GTP». Первым этапом синтеза дофамина является GTP циклогидролаза, и значительная недостаточность этого шага вызывает синдром, имеющий невропатологию, аналогичную LNS. Таким образом, отсутствие HGPRT может вызвать нуклеотидная недостаточность (в частности: дефицит GTP) расстройство, приводящее к дефициту дофамина.[14]
Было высказано предположение, что другая животная модель LNS возникла в результате окислительного повреждения, вызванного гиперурикемия сопровождающий LNS. Это основано на теории, что мочевая кислота это мощный Восстановитель и вероятно важный человек антиоксидант, в высокой концентрации в крови. Таким образом, было высказано предположение, что свободные радикалы, окислительный стресс, и активные формы кислорода может играть некоторую роль в невропатологии LNS.[12][15][16][неосновной источник необходим ]
Тем не менее, некоторые данные свидетельствуют против роли мочевой кислоты в нейропатологии синдрома Леша-Найхана:
- Гиперурикемия, связанная с классической первичная подагра, что вызвано низким содержанием мочевой кислоты почечный клиренс а не перепроизводство мочевой кислоты, не связано с невропатологией.
- Гипоурикемия возникает при ряде пуриновых заболеваний, в частности ксантинурия. Несмотря на полное отсутствие мочевой кислоты в крови, пациенты с ксантинурией не имеют каких-либо невропатологий или каких-либо других болезненных состояний, кроме камней в почках, вызванных накоплением нерастворимого ксантина вместо мочевой кислоты.[17]
Точно так же мочевая кислота плохо проникает через гематоэнцефалический барьер. Однако сейчас считается, что окислительный стресс из-за мочевой кислоты метаболический синдром, атеросклероз, и Инсульт, все синдромы, связанные с высоким уровнем мочевой кислоты. По аналогии, Супероксиддисмутаза («SOD») и SOD-миметики, такие как TEMPOL облегчить последствия гиперурикемии. Точно так же 6-гидроксидофамин (предполагаемая животная модель нейропатии Леша – Найхана), по-видимому, действует как нейротоксин, генерируя активные формы кислорода. Возможно, что окислительный стресс, вызванный другим оксипурином, таким как ксантин, вызывает заболевание.[нужна цитата ]
Диагностика
Когда у пораженного человека полностью развиваются три клинических элемента: гиперпродукция мочевой кислоты, неврологическая дисфункция, когнитивные и поведенческие нарушения, диагноз LNS ставится легко. Диагностировать сложнее на ранних стадиях, когда три особенности еще не очевидны. Признаки самоповреждающего поведения (SIB), результаты анализа родословных и новая молекулярная биология с генетическим тестированием (так называемая Диагностическая триада для LNS) часто подтверждают диагноз.[18] Подозрение часто возникает, когда задержка развития человека связана с гиперурикемией. В противном случае диагноз должен быть заявлен, если задержка развития связана с камнями в почках (нефролитиаз ) или кровь в моче (гематурия ), вызванные камнями мочевой кислоты. По большей части, синдром Леша – Найхана впервые подозревается при развитии поведения, связанного с нанесением самому себе травмы. Однако самоповреждающее поведение проявляется и в других условиях, в том числе неспецифических. Интеллектуальная недееспособность, аутизм, Синдром Ретта, Синдром Корнелии де Ланге, синдром Туретта, семейная дисавтономия, хореоакантоцитоз, сенсорная невропатия включая наследственную сенсорную невропатию 1 типа и несколько психических заболеваний. Из них только люди с синдромом Леша – Найхана, синдромом де Ланге и семейной дизавтономией рецидивируют как следствие потери ткани. Прикусывание пальцев и губ является определяющим признаком синдрома Леша – Найхана; при других синдромах, связанных с членовредительством, поведение обычно состоит из ударов головой и неспецифических членовредительства, но не укусов щек, губ и пальцев. Синдром Леша – Найхана следует четко рассматривать только тогда, когда самоповреждающее поведение имеет место в сочетании с гиперурикемией и неврологической дисфункцией.[нужна цитата ]
Диагностический подход
Урат креатинин (продукт распада креатинфосфата в мышцах) соотношение концентраций в моче повышено. Это хороший показатель перепроизводства кислоты. У детей младше десяти лет с LNS соотношение уратов и креатинина обычно превышает два. Круглосуточная экскреция уратов более 20 мг / кг также типична, но не диагностический. Гиперурикемия (концентрация мочевой кислоты в сыворотке> 8 мг / дл) часто присутствует, но недостаточно надежна для диагностики. Активность фермента HGPRT в клетки из любого типа ткань (например., кровь, культивированный фибробласты, или же лимфобласты ), составляющая менее 1,5% от нормальной активности фермента, подтверждает диагноз синдрома Леша – Найхана. Молекулярно-генетические исследования мутаций гена HPRT могут подтвердить диагноз и особенно полезны для последующего «тестирования на носительство» у женщин из группы риска, таких как близкие родственники по женской линии.[нужна цитата ]
Тестирование
Использование биохимический Тестирование на обнаружение носителей технически сложное и используется нечасто. Биохимические анализы, которые были выполнены на луковицах волос у женщин из группы риска, имели небольшое количество обоих ложный положительный результат и ложноотрицательные результаты. Если для тестирования на мутацию доступна только самка с подозрением на носительство, возможно, ее целесообразно вырастить. лимфоциты в 6-тиогуанине (a аналог пурина ), что позволяет выжить только клеткам с дефицитом HGPRT. Частота мутантов 0,5–5,0 × 10−2 встречается у самок-носителей, в то время как самка-не-носитель имеет частоту 1–20 × 10−6. Эта частота обычно сама по себе является диагностической.
Молекулярно-генетическое тестирование - наиболее эффективный метод тестирования, поскольку HPRT1 - единственный известный ген, связанный с LNS. Лица, отображающие полное значение Lesch – Nyhan фенотип у всех есть мутации в гене HPRT1. Анализ последовательности мРНК доступен клинически и может использоваться для обнаружения мутаций HPRT1 у мужчин, страдающих синдромом Леша – Найхана. Такие методы, как ОТ-ПЦР, мультиплексная геномная ПЦР и анализ последовательностей (кДНК и геномная ДНК), используемые для диагностики генетических заболеваний, выполняются на исследовательской основе. Если тесты RT-PCR приводят к тому, что кДНК показывает отсутствие целого экзон или экзоны, затем проводят мультиплексную геномную ПЦР. Мультиплексное геномное ПЦР-тестирование амплифицирует девять экзонов гена HPRT1 в виде восьми продуктов ПЦР. Если рассматриваемый экзон удален, соответствующая полоса будет отсутствовать в мультиплексной ПЦР. Однако, если экзон присутствует, экзон секвенируется для идентификации мутации, что приводит к исключению экзона из кДНК. Если с помощью ОТ-ПЦР не создается кДНК, то выполняется мультиплексная ПЦР с учетом того, что большая часть или весь ген уничтожен.[нужна цитата ]
Уход
Лечение LNS симптоматическое. Подагра лечится аллопуринол контролировать чрезмерное количество мочевая кислота. Камни в почках можно лечить литотрипсия, метод разбивания камней в почках с помощью ударных волн или лазерных лучей. Стандартного лечения неврологических симптомов LNS не существует. Некоторых можно облегчить с помощью лекарств карбидопа /леводопа, диазепам, фенобарбитал, или же галоперидол.[4]
Важно контролировать перепроизводство мочевой кислоты, чтобы снизить риск нефропатия, нефролитиаз и подагрический артрит. Наркотик аллопуринол используется для остановки преобразования оксипурины в мочевую кислоту и предотвратить развитие последующих артритные тофусы (возникает после хронической подагры), камни в почках, и нефропатия, вызванное заболеванием почек. Аллопуринол принимают внутрь в обычной дозе 3–20 мг / кг в день. Затем дозу корректируют, чтобы снизить уровень мочевой кислоты до нормального диапазона (<3 мг / дл). Большинство больных можно лечить аллопуринолом на протяжении всей жизни.
Никакие лекарства не эффективны для контроля экстрапирамидный двигательные особенности болезни. Спастичность тем не менее, может быть уменьшено введением баклофен или же бензодиазепины.
Ранее не существовало эффективных методов лечения нейроповеденческих аспектов заболевания. Даже у детей, получавших с рождения аллопуринол, развиваются поведенческие и неврологические проблемы, несмотря на то, что у них никогда не было высоких концентраций мочевой кислоты в сыворотке крови. Самоповреждением и другим поведением лучше всего справиться с помощью комбинации медицинских, физических и поведенческих вмешательств. Самоповреждения часто уменьшаются с помощью ограничителей. Шестьдесят процентов людей удалили зубы[нужна цитата ] во избежание членовредительства, что семьи считают эффективным методом управления.[нужна цитата ] Поскольку стресс увеличивает членовредительство, поведенческое управление с помощью техник отвращения (которые обычно снижают самоповреждение) фактически увеличивает самоповреждение у людей с LNS. Почти все пострадавшие нуждаются в ограничениях для предотвращения членовредительства, и их удерживают более 75% времени. Часто это делается по их собственному желанию и иногда включает ограничения, которые могут показаться неэффективными, поскольку они физически не предотвращают укусы. Семьи сообщают, что пострадавшие чувствуют себя более непринужденно, когда их сдерживают.
Медицинский и образовательный центр Матени [2] в Пипаке, штат Нью-Джерси,[когда? ] шесть пациентов с синдромом Леша – Найхана, которые считаются наибольшей концентрацией случаев LNS в одном месте и признаны ведущим источником информации по вопросам ухода.
Лечение пациентов с LNS, по словам Гэри Эдди, доктора медицины, медицинского директора[требуется разъяснение ], должны включать: 1) разумное использование защитных устройств; 2) использование поведенческой техники, обычно называемой «избирательное игнорирование», с перенаправлением действий; и 3) Редкое употребление лекарств.[нужна цитата ]
Статья в номере журнала от 13 августа 2007 г. Житель Нью-Йорка журнал, написанный Ричард Престон, обсуждает "стимуляция глубокого мозга «в качестве возможного лечения. Это было выполнено нескольким пациентам с синдромом Леша-Найхана доктором Такаоми Тайра в Токио и группой во Франции под руководством доктора Филиппа Куба. У некоторых пациентов наблюдалось уменьшение спастических самоповреждающих симптомов . Методика разработана для лечения людей с болезнь Паркинсона, по словам Престона, более 20 лет назад. Лечение включает в себя инвазивную операцию по размещению проводов, которые проводят постоянный электрический ток в определенной области мозга.[19]
Обнадеживающим достижением в лечении нейроповеденческих аспектов LNS стала публикация в номере журнала за октябрь 2006 г. Журнал наследственных метаболических заболеваний экспериментальной терапии пероральными S-аденозил-метионин (Одно и тоже).[20]Этот препарат нуклеотид предшественник который обеспечивает легко усваиваемый пурин, который, как известно, транспортируется через гематоэнцефалический барьер. Было показано, что введение SAMe взрослым пациентам с LNS улучшает нейроповеденческие и другие неврологические признаки. Препарат отпускается без рецепта и широко применяется при депрессия, но его использование для лечения LNS должно проводиться только под строгим медицинским наблюдением, поскольку побочные эффекты известны.
SAMe также недавно использовался для лечения другой пуриновой нуклеотидной болезни, «синдрома Арта» (который является расстройством PRPP, общим с LNS), с обнадеживающими результатами.[21]Таким образом, SAMe может быть полезен для лечения болезней пуриновых нуклеотидов, которые включают LNS.
Прогноз
Прогноз для людей с тяжелой формой LNS плохой. Смерть обычно происходит из-за почечная недостаточность или осложнения от гипотонии в первом или втором десятилетии жизни. Менее тяжелые формы есть лучшие прогнозы.[4]
История
Майкл Леш был студентом-медиком в Джонс Хопкинс и Уильям Найхан, а педиатр и биохимический генетик, был его наставником, когда эти двое определили LNS и связанную с ней гиперурикемию у двух больных братьев в возрасте 4 и 8 лет.[22] Леш и Найхан опубликовали свои выводы в 1964 году.[23] В течение трех лет метаболическая причина была идентифицирована Дж. Эдвин Сигмиллер и его коллеги по Национальные институты здравоохранения США.[24]
Рекомендации
- ^ Джеймс, Уильям Д .; Бергер, Тимоти Дж .; и другие. (2006). Кожные болезни Эндрюса: клиническая дерматология. Saunders Elsevier. п. 546. ISBN 978-0-7216-2921-6.
- ^ а б Синдром Леша – Найхана. Домашний справочник по генетике. Проверено 24 мая 2007.
- ^ Оле Даниэль Энерсен. Синдром Леша-Найхана в Кто это назвал?
- ^ а б c lesch_nyhan в NINDS
- ^ Хладник У., Нихан У.Л., Бертелли М (сентябрь 2008 г.). «Вариабельная экспрессия дефицита HPRT у 5 членов семьи с одной и той же мутацией». Arch. Neurol. 65 (9): 1240–3. Дои:10.1001 / archneur.65.9.1240. PMID 18779430.
- ^ Кауэлс Р.Г., Мартенс Л.К. (2005). «Самоуничтожение при синдроме Леша – Найхана». J. Oral Pathol. Med. 34 (9): 573–5. Дои:10.1111 / j.1600-0714.2005.00330.x. PMID 16138897.
- ^ а б Гуалтьери, К. Томас (2002). Травма головного мозга и умственная отсталость: психофармакология и нейропсихиатрия, п. 257. Lippincott Williams & Wilkins. ISBN 0-7817-3473-8. [1]
- ^ Augoustides-Savvopoulou P, Papachristou F, Fairbanks LD, Dimitrakopoulos K, Marinaki AM, Simmonds HA (2002). «Частичный дефицит гипоксантин-гуанинфосфорибозилтрансферазы как неожиданная причина почечного заболевания трех поколений: поучительная история». Педиатрия. 109 (1): E17. Дои:10.1542 / peds.109.1.e17. PMID 11773585.
- ^ Синдром Леша – Найхана. NCBI Гены и болезни. Проверено 12 апреля 2007 г.
- ^ Проктор, П. (26 декабря 1970 г.). «Побочные эффекты леводопы и синдром Леша-Нихана». Ланцет. 296 (7687): 1367. Дои:10.1016 / S0140-6736 (70) 92399-8. PMID 4098945.
- ^ Нихан WL (2000). «Функция дофамина при болезни Леша – Найхана». Environ. Перспектива здоровья. 108 (Дополнение 3): 409–11. Дои:10.2307/3454529. JSTOR 3454529. ЧВК 1637829. PMID 10852837.
- ^ а б Виссер Дж., Смит Д., Мой С., Бриз Дж., Фридман Т., Ротштейн Дж., Джинна Х. (2002). «Окислительный стресс и дефицит дофамина в генетической мышиной модели болезни Леша-Найхана». Brain Res Dev Brain Res. 133 (2): 127–39. Дои:10.1016 / S0165-3806 (02) 00280-8. PMID 11882343.
- ^ Бриз Г.Р., Кнапп DJ, Крисуэлл Х.Э., Мой С.С., Пападеас С.Т., Блейк Б.Л. «Новорожденная-6-гидроксидофамин-пораженная крыса: модель для клинической нейробиологии и нейробиологических принципов». Brain Res. Brain Res. Rev. 48 (1): 57–73. Дои:10.1016 / j.brainresrev.2004.08.004. PMID 15708628. S2CID 22599841.
- ^ Deutsch SI; Длинный КД; Россе РБ; Mastropaolo J; Эллер Дж. (Январь – февраль 2005 г.). «Предполагаемый дефицит пуринов на основе гуанина может способствовать аномалиям развития нервной системы, нейромодуляции и нейротрансмиссии при синдроме Леша-Найхана». Clin. Нейрофармакол. 28 (1): 28–37. Дои:10.1097 / 01.wnf.0000152043.36198.25. PMID 15711436. S2CID 36457793.
- ^ Saugstad O, Marklund S (1988). «Высокая активность глутатионпероксидазы эритроцитов у пациентов с синдромом Леша – Найхана». Acta Med Scand. 224 (3): 281–5. Дои:10.1111 / j.0954-6820.1988.tb19374.x. PMID 3239456.
- ^ Бавареско К., Кьярани Ф., Мэтте С., Вайнер М., Нетто С., де Соуза Вайз А (2005). «Влияние гипоксантина на активность Na +, K + -АТФазы и некоторые параметры окислительного стресса в полосатом теле крысы». Brain Res. 1041 (2): 198–204. Дои:10.1016 / j.brainres.2005.02.012. PMID 15829228. S2CID 22575382.
- ^ Кудо М; Мотеки Т; Сасаки Т; Konno Y; Ujiie S; Onose A; Mizugaki M; Ishikawa M; Хирацука М. (март 2008 г.). «Функциональная характеристика аллельных вариантов ксантиноксидазы человека». Pharmacogenet Genomics. 18 (3): 243–51. Дои:10.1097 / FPC.0b013e3282f55e2e. PMID 18300946. S2CID 8140455.
- ^ https://www.jstage.jst.go.jp/article/irdr/6/1/6_2016.01076/_pdf
- ^ Престон, Ричард (Август 2007 г.). «Ошибка в коде». Житель Нью-Йорка. п. 30. Получено 2008-09-14.
- ^ Глик Н. (октябрь 2006 г.). «Резкое снижение самоповреждений при болезни Леша – Найхана после введения S-аденозилметионина». J. Inherit. Метаб. Дис. 29 (5): 687. Дои:10.1007 / s10545-006-0229-8. PMID 16906475. S2CID 33099025.
- ^ де Брауэр А. П.; Williams KL; Duley JA; van Kuilenburg AB; Набуурс СБ; Эгмонт-Петерсен М; Lugtenberg D; Zoetekouw L; Запрещение MJ; Roeffen M; Hamel BC; Плетение L; Оврие Р.А.; Donald JA; Веверс Р.А.; Christodoulou J; ван Боховен Х. (сентябрь 2007 г.). «Синдром искусств вызван мутациями потери функции в PRPS1». Являюсь. J. Hum. Genet. 81 (3): 507–18. Дои:10.1086/520706. ЧВК 1950830. PMID 17701896.
- ^ Нихан WL (1997). «Признание синдрома Леша – Найхана врожденной ошибкой пуринового метаболизма» (PDF). J. Inherit. Метаб. Дис. 20 (2): 171–8. Дои:10.1023 / А: 1005348504512. PMID 9211189. S2CID 37373603.
- ^ Леш М., Нихан В. Л. (1964). «Семейное нарушение обмена мочевой кислоты и функции центральной нервной системы». Являюсь. J. Med. 36 (4): 561–70. Дои:10.1016/0002-9343(64)90104-4. PMID 14142409.
- ^ Сигмиллер JE, Rosenbloom FM, Келли WN (1967). «Дефект фермента, связанный с неврологическим расстройством человека, связанным с полом, и чрезмерным синтезом пуринов». Наука. 155 (770): 1682–4. Bibcode:1967Sci ... 155.1682S. Дои:10.1126 / science.155.3770.1682. PMID 6020292. S2CID 45609754.
внешняя ссылка
- Синдром Леша-Нихана в Национальные институты здравоохранения США Офис Редкие заболевания
- Статья GeneReview / NIH / UW о синдроме Леша – Найхана
- Национальный институт детского здоровья и развития человека (NICHD)
| Классификация | |
|---|---|
| Внешние ресурсы |