Синдром Каллмана - Kallmann syndrome
| Синдром Каллмана | |
|---|---|
| Другие имена | Наследственная аносмия Каллмана |
| Специальность | Эндокринология |
| Симптомы | Отсутствует или задерживается половое созревание, бесплодие, неспособность чувствовать запах |
| Осложнения | Остеопороз |
| Обычное начало | Присутствует при рождении |
| Продолжительность | На всю жизнь |
| Уход | Заместительная гормональная терапия Гонадотропин терапия |
| Частота | 1: 30 000 (мужчины), 1: 125 000 (женщины) |
Синдром Каллмана (KS) это генетический расстройство, которое мешает человеку начать или полностью завершить половое созревание. Синдром Каллмана - это форма группы состояний, называемых гипогонадотропный гипогонадизм. Чтобы отличить его от других форм гипогонадотропного гипогонадизма, синдром Каллмана имеет дополнительный симптом: полное отсутствие обоняния (аносмия) или снижение обоняния.[1][2][3] Если не лечить, люди будут плохо определять вторичные половые признаки, проявлять признаки гипогонадизм, почти всегда бесплодный и подвержены повышенному риску развития остеопороз.[1] Также может возникнуть ряд других физических симптомов, влияющих на лицо, руки и скелетную систему.[2]
Основная причина - сбой в правильном производстве или деятельности гонадотропин-рилизинг гормон посредством гипоталамус. Это приводит к снижению уровня половых гормонов. тестостерон у мужчин или эстроген и прогестерон у самок. Диагноз обычно ставится в подростковом возрасте, когда половое созревание не начинается.[3]
Обычно требуется пожизненное лечение для всех полов. Заместительная гормональная терапия (ЗГТ) - основная форма лечения, направленная на восполнение недостающего тестостерона или эстрогена и прогестерона. Также доступны специализированные процедуры для лечения бесплодия.[4]
Заболевание чаще диагностируется у мужчин, чем у женщин.[5] В 2011 году исследование финского населения показало, что заболеваемость составляет 1 случай из 48 000 человек, из которых 1 из 30 000 среди мужчин и 1 из 125 000 среди женщин.[6] Синдром Каллмана впервые был описан по имени в статье, опубликованной в 1944 г. Франц Йозеф Каллманн, а Немецкий -Американец генетик.[7][8] Связь между аносмией и гипогонадизмом уже была отмечена испанским врачом. Аурелиано Маэстре де Сан-Хуан в 1856 г.[9][10]
Признаки и симптомы


Обычно трудно отличить случай синдрома Каллмана (СК) / гипогонадотропного гипогонадизма (ГГ) от прямой конституциональной задержки половое созревание. Однако, если половое созревание не наступило ни к 14 годам (девочки), ни к 15 годам (мальчики) и присутствует один или несколько не репродуктивных признаков, упомянутых ниже, то направление к специалисту репродуктивный эндокринолог может быть целесообразным.[11][1][5]
Характеристики KS и других форм HH можно разделить на две разные категории; «репродуктивный» и «непродуктивный».[3][10][4][12][2]
Репродуктивные особенности
- Неспособность начать или полностью завершить половое созревание.[1]
- Отсутствие развития яичек у мужчин (размер <4 мл, тогда как нормальный диапазон составляет от 12 до 25 мл).[1]
- Начальный аменорея (не запускается менструация ).[5]
- Плохо выраженные вторичные половые признаки.[2]
- Микропенис в 5-10% случаев у мужчин.[1]
- Крипторхизм (неопущенные яички) при рождении.[1]
- Низкие уровни гонадотропины LH и ФСГ.[2]
- Гипогонадизм из-за низкого уровня тестостерон у мужчин или эстроген /прогестерон у женщин.[2]
- Бесплодие.[1]
Не репродуктивные особенности
- Полное отсутствие обоняния (аносмия ) или заметно сниженное обоняние (гипосмия). Это определяющая черта синдрома Каллмана; это не наблюдается в других случаях HH. Примерно 50% случаев ГГ происходят с аносмией и могут быть названы синдромом Каллмана.[2]
- Волчья пасть, заячья губа или другие срединные кранио-лицевые дефекты.[3]
- Нервное нарушение слуха[2]
- Отсутствие одной из почек (односторонняя агенезия почек)[2]
- Скелетные дефекты, включая расщепление кисти и стопы (эктродактилия ), укороченный средний палец (пястный)[2] или же сколиоз[13]
- Руководство синкинезия (зеркальные движения рук)[2]
- Отсутствие зубов (гиподонтия)[2]
- Плохой баланс или координация из-за церебральная атаксия.[5]
- Дефекты глаз, такие как колобома или же птоз.[10]
- Увеличение случаев дальтонизма [14][15]
Точная генетическая природа каждого конкретного случая KS / HH будет определять, какие из не репродуктивных особенностей, если таковые имеются, будут иметь место. Выраженность симптомов также будет варьироваться от случая к случаю. Даже у членов семьи не будет такого же диапазона или серьезности симптомов.[2][5]
KS / HH чаще всего присутствует с рождения, но версии с началом у взрослых встречаются как у мужчин, так и у женщин. В гипоталамо-гипофизарно-гонадная ось (Ось HPG) нормально функционирует при рождении и во взрослой жизни, обеспечивая нормальное половое созревание и нормальную репродуктивную функцию. Затем ось HPG либо полностью выходит из строя, либо снижается до очень низкого уровня высвобождения GnRH во взрослой жизни без очевидной причины (например, опухоль гипофиза). Это приведет к падению уровня тестостерона или эстрогена и бесплодию.[13][16]
Функциональная гипоталамическая аменорея наблюдается у женщин, где ось HPG подавляется в ответ на физический или психологический стресс или недоедание, но обратима при удалении фактора стресса.[1]
Некоторые случаи KS / HH, по-видимому, меняются в течение взрослой жизни, когда ось HPG возобновляет свою нормальную функцию, а уровни GnRH, LH и FSH возвращаются к нормальным уровням. Это происходит примерно у 10–22% людей, в первую очередь у нормосмических случаев ЗГГ, а не у пациентов с СК, и обнаруживается только у людей, которые прошли какую-либо форму заместительной терапии тестостероном. Обычно это обнаруживается только тогда, когда объем яичек увеличивается при лечении только тестостероном, а уровень тестостерона возвращается к норме после прекращения лечения. Этот тип KS / HH редко возникает в тех случаях, когда у мужчин в анамнезе не опускались яички.[5][3]
Люди, страдающие СК и другими формами ГГ, почти всегда рождаются с нормальной половой дифференциацией; т.е. они физически являются мужчинами или женщинами. Это связано с хорионическим гонадотропином человека (ХГЧ), продуцируемым плацента примерно от 12 до 20 недель беременность (беременность), на которую обычно не влияет KS или CHH.[17]
У людей с KS / HH отсутствует всплеск GnRH, LH и FSH, который обычно возникает между рождением и шестимесячным возрастом. Этот всплеск особенно важен для мальчиков, так как он помогает при опускании яичек в мошонку. Всплеск GnRH / LH / FSH у детей без KS / HH дает определяемые уровни тестостерона у мальчиков и эстрогена и прогестерона у девочек. Отсутствие этого всплеска иногда может быть использовано в качестве диагностического инструмента, если подозревается саркома-шевелюра / ГГ у новорожденного мальчика, но, как правило, оно недостаточно отчетливо для диагностики у девочек.[3]
Остеопороз
Одним из возможных побочных эффектов KS / CHH является повышенный риск развития вторичных остеопороз или же остеопения. Эстроген (женщины) или тестостерон (мужчины) необходимы для поддержания плотность костной ткани.[18] Дефицит тестостерона или эстрогена может увеличить скорость резорбция кости в то же время замедляя скорость формирование кости. В целом это может привести к ослаблению и хрупкости костей, которые имеют более высокую тенденцию к переломам.[нужна цитата ]
Даже непродолжительное время с низким уровнем эстрогена или тестостерона, поскольку в случаях поздней диагностики KS / CHH может привести к повышенному риску развития остеопороза, но при этом присутствуют и другие факторы риска, такие как курение, поэтому риск его развития будет варьироваться от человека к человеку. человек. Для контроля минеральной плотности костей рекомендуется сканирование плотности костей.[13]
Сканирование плотности костной ткани известно как двухэнергетическая рентгеновская абсорбциометрия сканирование (сканирование DEXA или DXA). Это простой тест, на выполнение которого требуется менее 15 минут. Это предполагает прохождение специализированного рентгеновский снимок изображение позвоночника и бедер, измерение минеральной плотности костей и сравнение результата со средним значением для молодого здорового взрослого в общей популяции.[19]
Адекватный кальций уровней и, наверное, что более важно, Витамин Д уровни необходимы для здоровой плотности костей. Некоторым людям с KS / CHH будут проверяться уровни, и им могут прописать дополнительные таблетки или инъекции витамина D, чтобы попытаться предотвратить ухудшение состояния. Роль витамина D для общего состояния здоровья в настоящее время находится под пристальным вниманием, и некоторые исследователи утверждают, что дефицит витамина D распространен во многих группах населения и может быть связан с другими заболеваниями.[20]
Некоторым людям с тяжелым остеопорозом могут быть назначены бисфосфонаты для сохранения костной массы в дополнение к заместительной гормональной терапии.[21]
Генетика
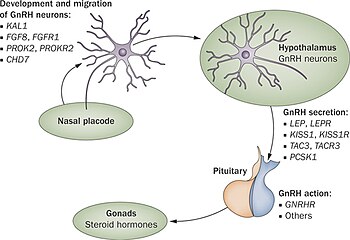
На сегодняшний день по меньшей мере 25 различных генов вовлечены в возникновение синдрома Каллмана или других форм гипогонадотропного гипогонадизма через нарушение производства или активности гонадолиберина (37). Эти задействованные гены охватывают все формы наследование и ни один дефект гена не является общим для всех случаев, что затрудняет генетическое тестирование и прогнозирование наследования.[22][23]
Число известных генов, вызывающих случаи KS / CHH, все еще увеличивается.[12] Кроме того, считается, что некоторые случаи KS / CHH вызваны двумя отдельными генными дефектами, возникающими одновременно.[5]
Дефекты отдельных генов могут быть связаны со специфическими симптомами, которые могут помочь в определении того, какие гены нужно проверить.[5][2] От 35 до 45% случаев KS / CHH имеют неизвестную генетическую причину.[24]
В ANOS1 дефект гена (ранее известный как KAL-1) был первым обнаруженным и наиболее часто тестируемым. Это вызывает х-связанный форма синдрома Каллмана и связана с дополнительными симптомами аносмия, бимануальный синкинезия и почечная агенезия. Считается, что этот дефект является причиной от 5 до 10% всех случаев синдрома Каллмана / ХГГ.[5][2]
Патофизиология
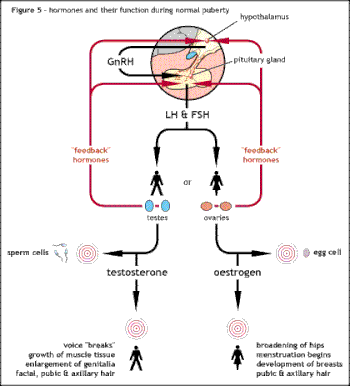

Основная причина синдрома Каллмана или других форм гипогонадотропного гипогонадизма - нарушение правильного действия гипоталамического гормона. ГнРГ. Термин изолированный дефицит гонадолиберина (IGD) все чаще используется для описания этой группы состояний, поскольку он подчеркивает основную причину этих состояний и отличает их от других состояний, таких как Синдром Клайнфельтера или же Синдром Тернера которые имеют сходные симптомы, но имеют другую этиологию.[25] Термин гипогонадизм описывает низкий уровень циркуляции крови. половые гормоны; тестостерон у мужчин и эстроген и прогестерон у самок. Гипогонадизм может возникать по разным причинам. Использование термина гипогонадотропный относится к тому факту, что гипогонадизм, обнаруживаемый при HH, вызван нарушением производства гонадотропин гормоны, обычно выделяемые передняя доля гипофиза известный как лютеинизирующий гормон (LH) и фолликулостимулирующего гормона (ФСГ).[12][24] В противном случае нарушение активности GnRH может быть связано с отсутствием нейронов, высвобождающих GnRH, внутри гипоталамуса. ГГ может возникать как изолированное состояние, при котором затрагивается только выработка ЛГ и ФСГ, или он может возникать в условиях комбинированного дефицита гипофиза.[нужна цитата ]
В первые 10 недель нормального эмбрионального развития нейроны, высвобождающие гонадолиберин, мигрируют из своего первоначального источника в носовую область и в конечном итоге попадают в гипоталамус. Эти нейроны берут свое начало в области развивающейся головы, обонятельная плакода, что даст начало обонятельному эпителию; затем они проходят через решетчатая пластина, вместе с волокнами обонятельных нервов и в ростральный передний мозг. Оттуда они мигрируют в то, что станет гипоталамусом. Любые проблемы с развитием волокон обонятельного нерва будут препятствовать продвижению нейронов, высвобождающих гонадолиберин, к мозгу.[26]
Диагностика
Диагностика KS и других форм CHH осложняется тем, что трудно различить нормальную конституциональную задержку полового созревания и случай KS / CHH.[27][4][28] Диагноз часто является одним из исключений, обнаруженных во время обследования задержка полового созревания.[29][30][31]
У мужчин использование соответствующих возрасту уровней тестостерона может помочь отличить случай KS / CHH от случая задержки полового созревания. Если полового созревания не наблюдается, особенно при отсутствии развития яичек, может потребоваться проверка эндокринолога-репродуктолога. Если половая зрелость не проявляется к 16 годам, человека следует направить на эндокринологическое обследование.[32] Постнатальная диагностика KS / CHH в возрасте до 6 месяцев иногда возможна, поскольку нормальный послеродовой гормональный всплеск гонадотропинов вместе с тестостероном или эстрогеном отсутствует у детей с KS / CHH. Этот недостаток обнаруживаемых гормонов в крови может использоваться как диагностический индикатор, особенно у младенцев мужского пола.[33]
У женщин диагностика иногда откладывается, так как другие причины аменорея Обычно перед рассмотрением дела о KS / CHH необходимо сначала провести расследование.[34]
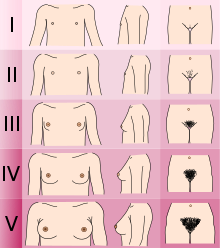
Диагностика нормального KS / CHH включает ряд клинических, биохимических и радиологических тестов, чтобы исключить другие состояния, которые могут вызывать подобные симптомы.[нужна цитата ]
Клинические испытания
- Сравнение роста со стандартными диаграммами роста.
- Определение Таннер этап полового развития. (Мужчины с KS / CHH обычно находятся на стадии I или II с развитием гениталий, женщины на стадии I с развитием груди, а мужчины и женщины - на стадии III с развитием лобковых волос).[2]
- Проверка на микропенис и неопустившиеся яички (крипторхизм ) у мужчин.
- Измерение объема яичка.
- Проверка развития груди и возраста в менархе у самок.
- Проверка обоняния с помощью панели одоранта или Тест на определение запаха Пенсильванского университета (UPSIT)
- Проверка на нарушение слуха.
- Проверка на отсутствие зубов или наличие заячья губа и / или волчья пасть.
- Проверка на пигментацию кожи и волос.
- Проверка зеркальных движений рук или признаков задержка нервного развития.
Лабораторные тесты
- Гормональное тестирование ранним утром, включая: ФСГ, LH, тестостерон, эстроген и пролактин.
- Тест на стимуляцию ГнРГ и / или ХГЧ для определения активности гипоталамус и гипофиз.
- Тест спермы
- Функция печени, функция почек и маркер воспаления тестирование.
- Кариотип для проверки на хромосомные аномалии.
Медицинская визуализация
- Выполнение запястья рентгеновский снимок для определения костного возраста.
- Мозг МРТ чтобы исключить любые структурные аномалии в гипоталамус или же гипофиз и проверить наличие обонятельные луковицы.
- УЗИ почек для исключения одностороннего почечная агенезия.
- Сканирование плотности костной ткани (DXA ) для проверки остеопороз или же остеопения.
Уход
Как для мужчин, так и для женщин первоначальной целью лечения является развитие вторичные половые признаки обычно наблюдается в период полового созревания.[2][35][30][31][36] Как только это будет достигнуто, продолжение заместительная гормональная терапия требуется как мужчинам, так и женщинам для поддержания половой функции, здоровья костей, либидо и общее благополучие.[3] У мужчин заместительная терапия тестостероном необходима для поддержания нормальной мышечной массы.[2]
Иногда требуется раннее лечение младенцев мужского пола с подозрением на KS / CHH для коррекции неопущенных яичек и микропенис если присутствует при использовании или хирургии или гонадотропин или же DHT лечение. Женщины с KS / CHH обычно не нуждаются в лечении до подросткового возраста. В настоящее время не существует лечения отсутствия обоняния, зеркального движения рук или отсутствия одной почки.[3]
Лечение как мужчин, так и женщин с KS / CHH обычно состоит из одного из трех вариантов, которые могут использоваться как для заместительной гормональной терапии, так и для лечения бесплодия.[2][3]
- Замещение половых гормонов (тестостерон или эстроген и прогестерон).
- Гонадотропная терапия (препараты, которые воспроизводят активность ФСГ и ЛГ).
- Пульсирующая терапия ГнРГ.
Заместительная гормональная терапия
Метод и доза лечения будут варьироваться в зависимости от человека, которого лечат. Начальное лечение обычно проводится с более низкими дозами у более молодых пациентов, чтобы развить вторичные половые признаки до того, как будут достигнуты дозы для взрослых.[2]
Для мужчин с KS / CHH типы доставки тестостерона включают ежедневные пластыри, ежедневное использование геля, ежедневные капсулы, подкожные или внутримышечные инъекции или шестимесячные имплантаты. Используются различные формы тестостерона, чтобы обеспечить как анаболический и андрогенный эффекты тестостерона достигаются.[3][4] Носовой Были разработаны методы доставки тестостерона, но их использование при лечении KS / CHH официально не оценивалось.[2]
Гонадотропиновая терапия в виде хорионический гонадотропин человека (ХГЧ) инъекции с использованием или без использования ФСГ также могут использоваться пациентам мужского пола для индукции развития вторичных половых признаков наряду с возможным индуцированием фертильности.[3]
Для женщин заместительная гормональная терапия предполагает использование эстрогена и прогестерона. Сначала эстроген используется в форме таблеток или геля для максимального развития груди, затем используется комбинация эстрогена и прогестерона.[3][2] Циклический прогестерон обычно необходим для поддержания эндометрий (подкладка матка ) здоровый.[2]
У мужчин наблюдение за лечением обычно требует измерения уровня тестостерона в сыворотке крови. ингибин B, гематокрит и простатоспецифический антиген (PSA). Если используются инъекции, измеряются минимальные уровни, чтобы обеспечить достижение адекватного уровня тестостерона на протяжении всего цикла инъекции.[3]
У женщин мониторинг обычно состоит из измерения эстрогена, ФСГ, ЛГ, ингибин B и антимюллеров гормон (AMH).[3]
Стандартная заместительная гормональная терапия обычно не способствует фертильности ни у мужчин, ни у женщин, при этом у мужчин не наблюдается роста яичек. Раннее лечение в подростковом возрасте может помочь в психологическом благополучии людей с KS / CHH.[3]
Лечение бесплодия
Терапия гонадотропинами может использоваться как для мужчин, так и для женщин, чтобы добиться фертильности у некоторых людей.[3][2]
Пульсирующая терапия гонадолиберином также может использоваться для стимуляции фертильности, особенно у женщин, но ее применение ограничено несколькими специализированными лечебными центрами.[2]
У мужчин с KS / CHH бесплодие в первую очередь связано с отсутствием сперма производство в яички. Производство спермы может быть достигнуто либо за счет использования гонадолиберина, вводимого с помощью микроинфузионного насоса, либо за счет инъекций гонадотропина (ХГЧ, ФСГ, чМГ ). Время, необходимое для достижения адекватного производства спермы для естественного зачатия, будет варьироваться от человека к человеку. Если до лечения яички очень маленькие и в анамнезе не опускались яички, может потребоваться больше времени для получения спермы. В этих случаях, вспомогательные репродуктивные технологии, например, получение спермы с использованием извлечение спермы из яичек (TESE) и / или интрацитоплазматическая инъекция спермы (ИКСИ), может потребоваться.[37]
У женщин с KS / CHH бесплодие в первую очередь связано с отсутствием созревание яиц, находящихся в яичники. Стимуляция овуляции может быть достигнута либо с помощью пульсирующей терапии гонадолиберином, либо с помощью инъекций гонадотропина (ХГЧ, ФСГ, чМГ), вводимых через определенные интервалы, чтобы вызвать созревание и высвобождение яйца для естественного зачатия.[37]
Прогноз
Об исчезновении симптомов сообщалось от 10% до 22% случаев.[38][2]
Случаи обратного развития наблюдались как при KS, так и при нормосмическом CHH, но, по-видимому, менее распространены в случаях KS (где также нарушается обоняние). Обратное изменение не всегда является постоянным, и точные генетические причины еще не полностью поняты.[39]
Эпидемиология
Эпидемиология синдрома Каллмана до конца не изучена. Отдельные исследования включают отчет 1986 года с обзором медицинских карт в сардинской армии, который показал распространенность 1 из 86000 мужчин.[40] и отчет Финляндии за 2011 год, в котором обнаружена распространенность 1: 30 000 для мужчин и 1: 125 000 для женщин.[41]
Синдром Каллмана встречается примерно в 4 раза чаще у мужчин, чем у женщин, но только в 2,5 раза чаще встречается у мужчин в семейных случаях.[40][41]
История

Синдром Каллмана впервые был описан по имени в статье, опубликованной в 1944 г. Франц Йозеф Каллманн, а Немецкий -Американец генетик.[7][8] Связь между аносмией и гипогонадизмом уже была отмечена испанским врачом. Аурелиано Маэстре де Сан-Хуан в 1856 г.[9] В 1950-х годах Де Морсье и Готье сообщили о частичном или полном отсутствии обонятельная луковица в мозгах мужчин с гипогонадизмом.[42][10]
Терминология
Терминология, используемая при описании случаев ДГ, различается и может включать:
- Дефицит гонадолиберина
- врожденный гипогонадотропный гипогонадизм (CHH)[43]
- идиопатический /изолированный гипогонадотропный гипогонадизм (IHH)
- нормосмический гипогонадотропный гипогонадизм (nHH)
- гипоталамический гипогонадизм
- ольфакто-генитальный синдром
Исследование
Кисспептин представляет собой белок, который регулирует высвобождение GnRH из гипоталамуса, который, в свою очередь, регулирует высвобождение LH и, в меньшей степени, FSH из передней доли гипофиза. Кисспептин и связанный с ним рецептор KISS1R как известно, участвуют в регуляции полового созревания. Исследования показали, что кисспептин может использоваться в диагностике и лечении некоторых случаев синдрома Каллмана и хронического гепатита.[44][45]
Рекомендации
- ^ а б c d е ж грамм час я «Синдром Каллмана». Домашний справочник по генетике. Медицинская библиотека США. Национальные институты здоровья. Информация о генетических и редких заболеваниях. 26 июня 2016 г.. Получено 17 декабря, 2017.
- ^ а б c d е ж грамм час я j k л м п о п q р s т ты v ш Икс у z аа ab ac Баласубраманиан Р., Кроули В. Ф. младший (2017). «Изолированный дефицит гонадотропин-высвобождающего гормона (ГнРГ)». SourceGeneReviews. PMID 20301509.
- ^ а б c d е ж грамм час я j k л м п о п q р s Бём У., Булу П.М., Даттани М.Т., де Ру Н., Доде С., Дункель Л., Дуайер А.А., Джакобини П., Харделин Дж. П., Юул А., Магни М., Питтелуд Н., Превот В., Райвио Т., Тена-Семпере М., Куинтон Р. , Янг Дж (сентябрь 2015 г.). «Экспертный консенсусный документ: Европейское консенсусное заявление о врожденном гипогонадотропном гипогонадизме - патогенез, диагностика и лечение». Обзоры природы. Эндокринология. 11 (9): 547–64. Дои:10.1038 / nrendo.2015.112. PMID 26194704.
- ^ а б c d Дункель Л., Куинтон Р. (июнь 2014 г.). «Переход в эндокринологии: индукция полового созревания». Европейский журнал эндокринологии. 170 (6): R229–39. Дои:10.1530 / EJE-13-0894. PMID 24836550.
- ^ а б c d е ж грамм час я Лима Амато LG, Latronico AC, Gontijo Silveira LF (июнь 2017 г.). «Молекулярно-генетические аспекты врожденного изолированного гипогонадотропного гипогонадизма». Клиники эндокринологии и метаболизма Северной Америки. 46 (2): 283–303. Дои:10.1016 / j.ecl.2017.01.010. PMID 28476224.
- ^ Лайтинен Э.М., Варалахти К., Томмиска Дж., Эклунд Э., Терваниеми М., Валанн Л., Райвио Т. (июнь 2011 г.). «Заболеваемость, фенотипические особенности и молекулярная генетика синдрома Каллмана в Финляндии». Журнал редких заболеваний Orphanet. 6 (17 июня): 41. Дои:10.1186/1750-1172-6-41. ЧВК 3143089. PMID 21682876.
- ^ а б Каллманн Ф. Дж., Шенфельд В. А., Баррера С. Е. (1943–1944). «Генетические аспекты первичного евнухоидизма». Am J Ment Defic. 48: 203–236.
- ^ а б синд / 2549 в Кто это назвал?
- ^ а б Маэстре де Сан-Хуан, Аурелиано (1856). "Teratolagia: falta total de los nervios olfactorios con anosmia en un Individual en quien existia una atrofia congenita de los testiculos y miembro viril". El Siglo Médico. 3: 211–221.
- ^ а б c d Ким Ш. (декабрь 2015 г.). «Врожденный гипогонадотропный гипогонадизм и синдром Каллмана: прошлое, настоящее и будущее». Эндокринология и метаболизм. 30 (4): 456–66. Дои:10.3803 / EnM.2015.30.4.456. ЧВК 4722398. PMID 26790381.
- ^ МакКейб MJ, Bancalari RE, Dattani MT (февраль 2014 г.). «Диагностика и оценка гипогонадизма». Обзоры детской эндокринологии. 11 Дополнение 2 (фев): 214–29. PMID 24683946.
- ^ а б c Митчелл А.Л., Дуайер А., Питтелуд Н., Куинтон Р. (июль 2011 г.). «Генетические основы и вариабельное фенотипическое выражение синдрома Каллмана: к объединяющей теории». Тенденции в эндокринологии и метаболизме. 22 (7): 249–58. Дои:10.1016 / j.tem.2011.03.002. PMID 21511493. S2CID 23578201.
- ^ а б c «Синдром Каллмана». Редкие заболевания. Национальная организация по редким заболеваниям (NORD). 2012 г.. Получено 16 декабря, 2017.
- ^ Чопра Р., Чандер А., Джейкоб Дж. Дж. (Май 2012 г.). «Глаз как окно в редкие эндокринные нарушения». Индийский журнал эндокринологии и метаболизма. 16 (3): 331–8. Дои:10.4103/2230-8210.95659. ЧВК 3354836. PMID 22629495.
- ^ Джаффе MJ, Шеринс RJ, de Monasterio F (1989). Недостатки цветового зрения IX. Documenta Ophthalmologica Proceedings Series. Дордрехт: Спрингер. С. 201–207. Дои:10.1007/978-94-009-2695-0_24. ISBN 9789401077156.
- ^ «Синдром Каллмана». Национальные институты здоровья. Медицинская библиотека США. Домашний справочник по генетике. Декабрь 2017 г.. Получено 17 декабря, 2017.
- ^ Сперлинг, Марк (2014). Электронная книга по детской эндокринологии. Elsevier Health Sciences. п. 136. ISBN 9781455759736.
- ^ Guo CY, Jones TH, Eastell R (февраль 1997 г.). «Лечение изолированного гипогонадотропного гипогонадизма, влияющего на минеральную плотность костной ткани и метаболизм». Журнал клинической эндокринологии и метаболизма. 82 (2): 658–65. Дои:10.1210 / jc.82.2.658. PMID 9024272.
- ^ Лайтинен Э.М., Герой М., Вааралахти К., Томмиска Дж., Райвио Т. (август 2012 г.). «Минеральная плотность костной ткани, состав тела и метаболизм у пациентов с врожденным гипогонадотропным гипогонадизмом». Международный журнал андрологии. 35 (4): 534–40. Дои:10.1111 / j.1365-2605.2011.01237.x. PMID 22248317.
- ^ Wimalawansa SJ, Razzaque DM, Al-Daghri NM (декабрь 2017 г.). «Кальций и витамин D в здоровье человека: обман или реальность?». Журнал стероидной биохимии и молекулярной биологии. 16 декабря: 4–14. Дои:10.1016 / j.jsbmb.2017.12.009. PMID 29258769. S2CID 11467429.
- ^ Голдс Г., Хоудек Д., Арнасон Т. (2017). «Мужской гипогонадизм и остеопороз: эффекты, клинические последствия и лечение дефицита тестостерона для здоровья костей». Int J Endocrinol. 2017: 4602129. Дои:10.1155/2017/4602129. ЧВК 5376477. PMID 28408926.
- ^ Layman LC (май 2013 г.). «Клинические генетические исследования синдрома Каллмана». Журнал клинической эндокринологии и метаболизма. 98 (5): 1860–2. Дои:10.1210 / jc.2013-1624. ЧВК 3644595. PMID 23650337.
- ^ Вальдес-Социн Х, Рубио Альманса М, Томе Фернандес-Ладреда М, Дебрей Ф.Г., Бурс В., Бекерс А (2014). «Нарушения репродуктивной функции, обоняния и нервного развития: генетические дефекты при различных гипогонадотропных гипогонадальных синдромах». Границы эндокринологии. 5 (109): 109. Дои:10.3389 / fendo.2014.00109. ЧВК 4088923. PMID 25071724.
- ^ а б Vezzoli V, Duminuco P, Bassi I, Guizzardi F, Persani L, Bonomi M (июнь 2016 г.). «Сложная генетическая основа врожденного гипогонадотропного гипогонадизма». Минерва Эндокринологика. 41 (2): 223–39. PMID 26934720.
- ^ Au MG, Кроули В.Ф., Бак С.Л. (октябрь 2011 г.). «Генетическое консультирование при изолированном дефиците ГнРГ». Молекулярная и клеточная эндокринология. 346 (1–2): 102–9. Дои:10.1016 / j.mce.2011.05.041. ЧВК 3185214. PMID 21664415.
- ^ Тейшейра Л., Гимиот Ф., Доде С., Фалле-Бьянко С., Миллар Р.П., Делезоид А.Л., Харделин Дж. П. (октябрь 2010 г.). «Нарушение миграции нейроэндокринных клеток GnRH в арринэнцефалических условиях человека». Журнал клинических исследований. 120 (10): 3668–72. Дои:10.1172 / JCI43699. ЧВК 2947242. PMID 20940512.
- ^ Pitteloud N (декабрь 2012 г.). «Управление задержкой или измененным половым созреванием у мальчиков». BMJ. 345 (3 декабря): e7913. Дои:10.1136 / bmj.e7913. PMID 23207503. S2CID 5159169.
- ^ Янг Дж (март 2012 г.). «Обращение к пациенту мужского пола с врожденным гипогонадотропным гипогонадизмом». Журнал клинической эндокринологии и метаболизма. 97 (3): 707–18. Дои:10.1210 / jc.2011-1664. PMID 22392951.
- ^ Ли PA, Houk CP (13 августа 2012 г.). «Самый маленький ребенок в школе: оценка задержки полового созревания». Medscape Педиатрия.
- ^ а б Джонс H, изд. (2008). «Глава 9: Половое созревание и фертильность». Дефицит тестостерона у мужчин. Оксфордская эндокринологическая библиотека. ISBN 978-0199545131.
- ^ а б Jockenhovel F (2004). «Глава 3: Диагностика гипогонадизма». Мужской гипогонадизм. Uni-Med Science. ISBN 978-3-89599-748-8.
- ^ Quinton R (апрель 2005 г.). «Развитие подростков: советы по азбуке подросткового возраста могут ввести в заблуждение». BMJ. 330 (7494): 789, ответ автора 789. Дои:10.1136 / bmj.330.7494.789. ЧВК 555895. PMID 15802728.
- ^ Двайер А.А., Джаясена С.Н., Куинтон Р. (июнь 2016 г.). «Врожденный гипогонадотропный гипогонадизм: последствия отсутствия мини-полового созревания». Минерва Эндокринологика. 41 (2): 188–95. PMID 27213784.
- ^ Брай-Гуйяр, Трабадо С., Булиган Дж., Сарфати Дж., Франсу Б., Саленав С., Шансон П., Брайи-Табард С., Гиошон-Мантель А., Янг Дж. (Май 2010 г.). «Врожденный гипогонадотропный гипогонадизм у женщин: клинический спектр, оценка и генетика». Анналы д'Эндокринологии. 71 (3): 158–62. Дои:10.1016 / j.ando.2010.02.024. PMID 20363464.
- ^ Буваттье С., Майоне Л., Булиган Дж., Доде С., Гиошон-Мантель А., Янг Дж. (Октябрь 2011 г.). «Неонатальная терапия гонадотропинами при мужском врожденном гипогонадотропном гипогонадизме». Обзоры природы. Эндокринология. 8 (3): 172–82. Дои:10.1038 / nrendo.2011.164. PMID 22009162. S2CID 4564169.
- ^ Хан Т.С., премьер-министр Булу (июнь 2010 г.). «Какая оптимальная терапия для молодых мужчин с гипогонадотропным гипогонадизмом?». Клиническая эндокринология. 72 (6): 731–7. Дои:10.1111 / j.1365-2265.2009.03746.x. PMID 19912242.
- ^ а б Майоне Л., Дуайер А.А., Франсу Б., Гиошон-Мантель А., Бинарт Н., Булиганд Дж., Янг Дж. (Март 2018 г.). «ГЕНЕТИКА В ЭНДОКРИНОЛОГИИ: Генетическое консультирование при врожденном гипогонадотропном гипогонадизме и синдроме Каллмана: новые вызовы в эпоху олигогенизма и секвенирования следующего поколения». Европейский журнал эндокринологии. 178 (3): R55 – R80. Дои:10.1530 / EJE-17-0749. PMID 29330225.
- ^ Сидхум В.Ф., Чан Ю.М., Липпинкотт М.Ф., Баласубраманиан Р., Куинтон Р., Пламмер Л., Дуайер А., Питтелуд Н., Хейс Ф.Дж., Холл Дж. Э., Мартин К.А., Боппл П.А., Семинара С.Б. (март 2014 г.). «Обращение и рецидив гипогонадотропного гипогонадизма: устойчивость и хрупкость репродуктивной нейроэндокринной системы». Журнал клинической эндокринологии и метаболизма. 99 (3): 861–70. Дои:10.1210 / jc.2013-2809. ЧВК 3942233. PMID 24423288.
- ^ Двайер А.А., Райвио Т., Питтелуд Н. (июнь 2016 г.). «ЛЕЧЕНИЕ ЭНДОКРИННОЙ БОЛЕЗНИ: Обратимый гипогонадотропный гипогонадизм». Европейский журнал эндокринологии. 174 (6): R267–74. Дои:10.1530 / EJE-15-1033. PMID 26792935.
- ^ а б Тритос, Николай А (10 октября 2016 г.). «Синдром Каллмана и идиопатический гипогонадотропный гипогонадизм: история вопроса, патофизиология, эпидемиология». eMedicine. Цитировать журнал требует
| журнал =(помощь) - ^ а б Баласубраманиан Р., Кроули В.Ф. (2 марта 2017 г.). «Изолированный дефицит гонадотропин-высвобождающего гормона (ГнРГ)». GeneReviews. Вашингтонский университет, Сиэтл. PMID 20301509.
- ^ De Morsier G, Gauthier G (ноябрь 1963 г.). «[Олфакто-генитальная дисплазия]». Патология и биология. 11: 1267–72. PMID 14099201.
- ^ Вальдес-Социн Х, Рубио Альманса М, Томе Фернандес-Ладреда М, Дебрей Ф.Г., Бурс В., Бекерс А (2014). «Нарушения репродуктивной функции, обоняния и нервного развития: генетические дефекты при различных гипогонадотропных гипогонадальных синдромах». Границы эндокринологии. 5: 109. Дои:10.3389 / fendo.2014.00109. ЧВК 4088923. PMID 25071724.
- ^ Скорупскайте К, Джордж Дж. Т., Андерсон Р. А. (2014). «Путь кисспептин-ГнРГ в репродуктивном здоровье и болезнях человека». Обновление репродукции человека. 20 (4): 485–500. Дои:10.1093 / humupd / dmu009. ЧВК 4063702. PMID 24615662.
- ^ Джордж JT, Seminara SB (ноябрь 2012 г.). «Кисспептин и гипоталамический контроль репродукции: уроки человека». Эндокринология. 153 (11): 5130–6. Дои:10.1210 / en.2012-1429. ЧВК 3473216. PMID 23015291.
внешняя ссылка
| Классификация | |
|---|---|
| Внешние ресурсы |