Живая полимеризация - Living polymerization
В химия полимеров, живая полимеризация это форма полимеризация с ростом цепи где возможность роста полимерная цепь к прекратить был удален.[1][2] Это можно сделать разными способами. Завершение цепочки и реакции передачи цепи отсутствуют, а скорость цепное инициирование также намного больше, чем скорость распространение цепи. В результате полимерные цепи растут с более постоянной скоростью. ставка чем в традиционной сети полимеризация и их длина остается очень похожей (т.е. у них очень низкий индекс полидисперсности ). Живая полимеризация - популярный метод синтеза блок-сополимеры поскольку полимер можно синтезировать поэтапно, каждая стадия содержит разные мономер. Дополнительные преимущества предопределены молярная масса и контроль над конечные группы.
Живая полимеризация желательна, потому что она обеспечивает точность и контроль синтеза макромолекул. Это важно, поскольку многие новые / полезные свойства полимеров обусловлены их микроструктурой и молекулярной массой. С молекулярный вес и дисперсность менее контролируемы при неживой полимеризации, этот метод более желателен для дизайна материалов[3][4]
Во многих случаях реакции живой полимеризации путают или считают синонимом контролируемой полимеризации. Хотя эти реакции полимеризации очень похожи, существует явное различие в определениях этих двух реакций. В то время как «живые» полимеризации определяются как реакции полимеризации, в которых устраняется обрыв цепи или передача цепи, реакции контролируемой полимеризации - это реакции, в которых обрывание подавляется, но не устраняется, посредством введения полимера в неактивное состояние.[3][4] Однако это различие все еще обсуждается в литературе.
Основные методы живой полимеризации:
- Живая анионная полимеризация
- Живая катионная полимеризация
- Жизнь метатезис полимеризация с раскрытием цикла
- Живая свободнорадикальная полимеризация
- Поликонденсации с живой цепью роста
История
Живая полимеризация была продемонстрирована Майкл Шварц в 1956 г. при анионной полимеризации стирол с щелочной металл / нафталин система в тетрагидрофуран (THF). Шварц показал, что перенос электронов произошло из анион-радикал из нафталин к стирол. Исходный анион-радикал стирола превращается в дианион (или, что эквивалентно, дизоди-) разновидности, к которым быстро добавляется стирол с образованием «двустороннего живого полимера». Важным аспектом своей работы Шварц использовал апротонный растворитель тетрагидрофуран, который растворяется, но в остальном не реагирует с металлоорганическими промежуточными продуктами. После первоначального добавления мономера в систему инициатора вязкость будет увеличиваться (из-за увеличения роста полимерной цепи), но в конечном итоге прекратится после истощения концентрации мономера. Однако он обнаружил, что добавление более мономер вызывал увеличение вязкости, что указывало на рост полимерной цепи, и, таким образом, был сделан вывод о том, что полимерные цепи никогда не обрывались.[6] Это был важный шаг в химии полимеров, поскольку контроль за тем, когда полимер гасился или прекращался, обычно не был контролируемым шагом. С этим открытием список потенциальных приложений резко расширился.[7]
Сегодня живые полимеризации широко используются в производстве многих типов полимеров и пластиков. Подход предлагает управление химическим составом полимера и, следовательно, структурными и электронными свойствами материала. Такой уровень контроля редко существует в неживых реакциях полимеризации.[4][8]
Высокая скорость инициирования: низкая полидисперсность


Одной из ключевых характеристик живой полимеризации является то, что реакции обрыва цепи и переноса практически исключаются из четырех элементарных реакций полимеризация с ростом цепи оставив только инициация и (цепные) реакции распространения.
Ключевой характеристикой живой полимеризации является то, что скорость инициации (что означает, что спящие химические частицы генерируют активные виды, способствующие размножению цепи) намного выше, чем скорость распространения цепи. Таким образом, все цепи растут с одинаковой скоростью (скоростью распространения).
Высокая скорость инициирования (вместе с отсутствием обрыва) приводит к низкому (или узкому) индексу полидисперсности (PDI), что указывает на широту распределения полимерных цепей (Живые полимеры ) Увеличенный срок службы растущей цепи, позволяющий осуществлять образование сополимера и функционализацию концевых групп в живой цепи. Эти факторы также позволяют прогнозировать молекулярную массу, выраженную как среднечисловую молекулярную массу (Mп). Для идеальной живой системы предполагается, что эффективность генерации активных видов составляет 100%, где каждый инициатор генерирует только один активный вид. Кинетическая длина цепи (среднее количество мономеров, с которыми активный компонент реагирует в течение своего срока службы) в данный момент времени можно оценить, зная концентрацию оставшегося мономера. Среднечисловая молекулярная масса, Mп, линейно увеличивается с процентом конверсии во время живой полимеризации
![v = { frac {[M] _ {0} - [M]} {[I] _ {0}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/db304320640f163330c3130db03ff9691e1a8ffa)
Методы
Живая анионная полимеризация
Еще в 1936 г. Карл Циглер предположили, что анионная полимеризация стирола и бутадиена путем последовательного добавления мономера к алкиллитиевому инициатору происходит без передачи или обрыва цепи. Двадцать лет спустя Шварц продемонстрировал живую полимеризацию. анионная полимеризация из стирол в THF с помощью нафталинид натрия как инициатор.[9][6][10]
Анион нафталина инициирует полимеризацию, восстанавливая стирол до его анион-радикала, который димеризуется до дилитиодифенилбутана, который затем инициирует полимеризацию. Эти эксперименты основывались на способности Шварца контролировать уровни примесей, которые разрушили бы высокореакционные металлоорганические промежуточные соединения.
Полимеризация живого α-олефина
α-олефины может быть полимеризован посредством анионной координационной полимеризации, при которой металлический центр катализатора считается противокатионом для анионный конец алкильной цепи (через координацию M-R). Инициаторы Циглера-Натта были разработаны в середине 1950-х годов и представляют собой гетерогенные инициаторы, используемые при полимеризации альфа-олефинов. Эти инициаторы были не только первыми, кто получил поли (1-алкены) с относительно высокой молекулярной массой (в настоящее время самый широко производимый термопласт в мире.Полиэтилен ) и PP (Полипропилен )[11] но инициаторы также были способны к стереоселективной полимеризации, которую приписывают хиральный Кристальная структура гетерогенного инициатора.[4] Ввиду важности этого открытия Зиглеру и Натте были вручены Нобелевская премия по химии 1963 г.. Хотя активные частицы, образованные из инициатора Циглера-Натта, обычно имеют длительные сроки жизни (в масштабе часов или более), время жизни размножающихся цепей сокращается из-за нескольких путей передачи цепи (Удаление бета-гидрида и передача соинициатору) и в результате не считаются живыми.[4]
Металлоценовые инициаторы рассматриваются как разновидность инициаторов Циглера-Натта в связи с использованием двухкомпонентной системы, состоящей из переходный металл и соинициатор металла I-III групп (например, метилалюмоксан (МАО) или другие соединения алкилалюминия). В металлоцен инициаторы образуют единый однородный сайт катализаторы которые изначально были разработаны для изучения влияния, которое структура катализатора оказывает на структуру / свойства получаемых полимеров; что было сложно для многосайтовых гетерогенных инициаторов Циглера-Натта.[11] Благодаря дискретному единственному центру на металлоценовом катализаторе исследователи смогли настроить и связать, как структура вспомогательного лиганда (не участвующего непосредственно в химических превращениях) и симметрия относительно хирального металлического центра влияют на микроструктуру полимера.[12] Однако из-за реакций разрыва цепи (в основном отщепления бета-гидрида) известно очень мало полимеризации на основе металлоцена.[4]
Путем настройки стерического объема и электронных свойств вспомогательных лигандов и их заместители класс инициаторов, известный как хелат инициаторы (или постметаллоценовые инициаторы) успешно использовались для стереоспецифический живые полимеризации альфа-олефинов. Хелатные инициаторы обладают высоким потенциалом для живых полимеризаций, поскольку вспомогательные лиганды могут быть сконструированы так, чтобы препятствовать или ингибировать пути обрыва цепи. Хелатные инициаторы можно дополнительно разложить на вспомогательные лиганды; анса-циклопентиадиениламидо-инициаторы, альфа-дииминовые хелаты и фенокси-иминные хелаты.[4]
- Инициаторы анса-циклопентадиениламидо (CpA)

У инициаторов CpA есть один циклопентадиенил заместитель и один или несколько заместителей азота, координированных с металлическим центром (обычно Zr или Ti) (Odian). Ацетамидинат диметил (пентаметилциклопентил) циркония на рисунке ___ был использован для стереоспецифической живой полимеризации 1-гексена при -10 ° C. Полученный поли (1-гексен) был изотактический (стереохимия одинакова между соседними повторяющимися единицами) подтверждается 13C-ЯМР. Множественные испытания продемонстрировали управляемость и предсказуемость (от катализатора до мономер соотношение) Mп с низким Đ. Далее было подтверждено, что полимеризация протекает, последовательно добавляя 2 части мономера, вторую часть добавляли после того, как первая часть уже была полимеризована, и отслеживая Đ и Mп цепи. Полученные полимерные цепи соответствовали прогнозам Mп (с общей концентрацией мономера = часть 1 + 2) и показал низкую Đ[13] предполагая, что цепи все еще были активными или живыми, когда была добавлена вторая часть мономера (5).
- α-дииминовые хелатные инициаторы
α-дииминовые хелатные инициаторы характеризуются наличием диимин хелатирующая структура вспомогательного лиганда, которая обычно координируется с металлическим центром с поздним переходом (то есть Ni и Pd).

Brookhart et al. провел обширную работу с этим классом катализаторов и сообщил о живой полимеризации α-олефинов.[14] и продемонстрировали живые чередующиеся сополимеры α-олефина монооксида углерода.[15]
Живая катионная полимеризация
Мономеры для живой катионной полимеризации - это богатые электронами алкены, такие как виниловые эфиры, изобутилен, стирол, и N-винилкарбазол. Инициаторы представляют собой бинарные системы, состоящие из электрофила и кислоты Льюиса. Метод был разработан примерно в 1980 году при участии Хигашимуры, Савамото и Кеннеди. Обычно создание стабильного карбокатиона в течение длительного периода времени затруднено из-за возможности гашения катиона β-протонами, присоединенными к другому мономеру в основной цепи, или в свободном мономере. Поэтому используется другой подход.[3][4][16]

В этом примере карбокатион генерируется добавлением кислоты Льюиса (соинициатор вместе с галогеном «X», уже находящимся в полимере - см. Рисунок), что в конечном итоге приводит к образованию карбокатиона в слабом равновесии. Это равновесие в значительной степени способствует состоянию покоя, оставляя мало времени для постоянного гашения или прекращения другими путями. Кроме того, можно добавить слабый нуклеофил (Nu :), чтобы еще больше снизить концентрацию активных частиц, таким образом сохраняя полимер «живым».[3][4][16] Однако важно отметить, что по определению полимеры, описанные в этом примере, технически не являются живыми из-за введения неактивного состояния, поскольку прекращение только уменьшилось, а не устранено (хотя эта тема все еще обсуждается). Но они действительно работают аналогично и используются в приложениях, аналогичных приложениям настоящей живой полимеризации.
Полимеризация метатезиса с раскрытием живого кольца
При правильных условиях реакции метатезис полимеризация с раскрытием цикла (ROMP) можно сделать живым. Первые такие системы были описаны Роберт Х. Граббс в 1986 г. на основе норборнен и Реактив Теббе а в 1978 году Граббс вместе с Ричард Р. Шрок описывая живую полимеризацию с вольфрам карбеновый комплекс.[17]
Обычно реакции ROMP включают превращение циклического олефина со значительной кольцевой деформацией (> 5 ккал / моль), такого как циклобутен, норборнен, циклопентен и т.д., в полимер, который также содержит двойные связи. При метатезисной полимеризации с раскрытием цикла важно отметить, что двойная связь обычно сохраняется в основной цепи, что позволяет считать ее «живой» при правильных условиях.[18]
Чтобы реакция ROMP считалась «живой», необходимо соблюдать несколько правил:[18]
- Быстрое и полное инициирование мономера. Это означает, что скорость, с которой инициирующий агент активирует мономер для полимеризации, должна происходить очень быстро.
- Сколько мономеров составляет каждый полимер (степень полимеризации), должно быть линейно связано с количеством мономера, с которого вы начали.
- В дисперсность полимера должно быть <1,5. Другими словами, распределение длины полимерных цепей в реакции должно быть очень низким.
Принимая во внимание эти рекомендации, это позволяет вам создать полимер, который хорошо контролируется как по содержанию (какой мономер вы используете), так и по свойствам полимера (что в значительной степени можно отнести к длине цепи полимера). Важно отметить, что живые полимеризации с раскрытием кольца могут быть анионными. или же катионный.

Поскольку у живых полимеров была удалена их способность обрыва цепи, это означает, что после того, как ваш мономер израсходован, добавление большего количества мономера приведет к тому, что полимерные цепи будут продолжать расти, пока не будет израсходован весь дополнительный мономер. Это будет продолжаться до тех пор, пока металлический катализатор в конце цепи не будет намеренно удален путем добавления гасящего агента. В результате это потенциально может позволить создать блокировать или же градиентный сополимер довольно легко и точно. Это может привести к высокой способности настраивать свойства полимера в зависимости от желаемого применения (электрическая / ионная проводимость и т. Д.)[4][18]
«Живая» свободнорадикальная полимеризация
Начиная с 1970-х годов было открыто несколько новых методов, которые позволили разработать живую полимеризацию с использованием свободный радикал химия. Эти методы включали каталитическая передача цепи полимеризация, полимеризация, опосредованная инициатором, стабильная полимеризация, опосредованная свободными радикалами (SFRP), радикальная полимеризация с переносом атома (ATRP), обратимая передача цепи присоединения-фрагментации (РАФТ ) полимеризация и полимеризация с переносом йода.
В «живой» радикальной полимеризации (или контролируемой радикальной полимеризации (CRP)) пути разрыва цепи сильно подавлены по сравнению с традиционной радикальной полимеризацией (RP), и CRP может проявлять характеристики живой полимеризации. Однако, поскольку обрыв цепи не отсутствует, а только минимизирован, СРБ технически не соответствует требованиям, предъявляемым IUPAC к живой полимеризации (определение IUPAC см. Во введении). Этот вопрос является предметом обсуждения, точки зрения различных исследователей можно найти в специальном выпуске журнала «Наука о полимерах» под названием Жить или под контролем?. Этот вопрос еще не решен в литературе, поэтому его часто обозначают как «живая» полимеризация, квазивоживая полимеризация, псевдоживая и другие термины для обозначения этой проблемы.
В CRP используются две общие стратегии для подавления реакций разрыва цепи и содействия быстрому инициированию по сравнению с размножением. Обе стратегии основаны на установлении динамического равновесия между активным размножающимся радикалом и спящими видами.[19]
Первая стратегия включает механизм обратимого захвата, при котором распространяющийся радикал подвергается активации / дезактивации (т. Е. Радикальная полимеризация с переносом атома ) процесс с разновидностью X. Частица X является стойким радикалом или разновидностью, которая может генерировать стабильный радикал, который не может закончиться сам с собой или размножаться, но может только обратимо "закончиться" с распространяющимся радикалом (от растущей полимерной цепи) П*. P * - радикальный вид, способный размножаться (kп) и необратимо завершаются (kт) с другим P *. X обычно представляет собой нитроксид (т.е. ТЕМП используется в Радикальная полимеризация, опосредованная нитроксидом ) или металлорганический вид. Покоящиеся виды (Pп-X) может быть активирован для регенерации активных размножающихся видов (P *) спонтанно, термически, с использованием катализатора и оптически.[19][20]
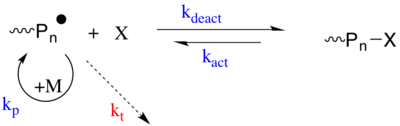
Вторая стратегия основана на дегенеративном переносе (DT) распространяющегося радикала между агентом переноса, который действует как спящие частицы (т. Е. Обратимая аддитивно-фрагментарная полимеризация с переносом цепи ). CRP на основе DT следуют традиционной кинетике радикальной полимеризации, то есть медленному инициированию и быстрому завершению, но агент переноса (Pm-X или Pn-X) присутствует в гораздо более высокой концентрации по сравнению с радикальным инициатором. Развивающиеся радикальные частицы подвергаются термически нейтральному обмену с неактивным агентом переноса посредством переноса атомов, переноса группы или химии присоединительных фрагментов.[19]

Поликонденсации с живой цепью роста
Поликонденсационная полимеризация с цепным ростом первоначально была разработана в предположении, что изменение замещающих эффектов полимера по отношению к мономеру приводит к тому, что концевые группы полимера становятся более реакционноспособными, что получило название «реакционная промежуточная поликонденсация». Существенный результат состоит в том, что мономеры предпочтительно реагируют с активированными концевыми группами полимера, а не с другими мономерами. Эта предпочтительная реакционная способность является фундаментальным различием при классификации механизма полимеризации как механизма роста цепи в отличие от ступенчатый рост в котором мономер и концевые группы полимерной цепи имеют равную реакционную способность (реакционная способность не контролируется). Несколько стратегий были использованы для минимизации реакций мономер-мономер (или самоконденсации), и полимеризация с низким D и контролируемым Mn была достигнута с помощью этого механизма для полимеров с небольшой молекулярной массой.[21] Однако для полимерных цепей с высокой молекулярной массой (то есть с малым соотношением инициатора к мономеру) Mn нелегко контролировать для некоторых мономеров, поскольку самоконденсация между мономерами происходит чаще из-за низкой концентрации размножающихся частиц.[21]
Поликонденсация с переносом катализатора
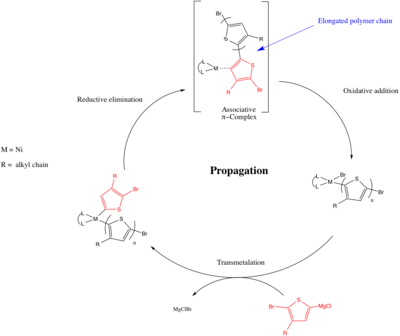
Поликонденсация с переносом катализатора (CTP) представляет собой механизм поликонденсации с ростом цепи, при котором мономеры не взаимодействуют напрямую друг с другом, а вместо этого мономер будет реагировать только с концевой группой полимера через механизм, опосредованный катализатором.[21] Общий процесс состоит из катализатора, активирующего концевую группу полимера, с последующей реакцией концевой группы со вторым входящим мономером. Затем катализатор переносится на удлиненную цепь при активации концевой группы (как показано ниже).[22]

Поликонденсация с переносом катализатора обеспечивает живую полимеризацию π-сопряженных полимеров и была открыта Цутому Йокозава в 2004 году.[22] и Ричард Маккалоу.[23] В CTP этап распространения основан на реакциях органического кросс-сочетания (т.е. Муфта Кумада, Муфта Соногашира, Муфта Негиши ) top образуют углерод-углеродные связи между дифункциональными мономерами. Когда Йокозава и Маккалоу независимо друг от друга открыли полимеризацию с использованием металлического катализатора для соединения Реактив Гриньяра с галогенидом, образующим новую углерод-углеродную связь. Механизм ниже показывает образование поли (3-алкилтиофена) с использованием инициатора Ni (Lп возможно 1,3-бис (дифенилфосфино) пропан (dppp) ) и аналогичен обычному механизму для Муфта Кумада с участием окислительная добавка, а трансметалляция и восстановительное устранение шаг. Однако есть ключевое различие: после восстановительного элиминирования в CTP образуется ассоциативный комплекс (который был подтвержден экспериментами по конкуренции внутри- / межмолекулярного окислительного присоединения.[8]), и последующее окислительное присоединение происходит между металлическим центром и связанной цепью (внутримолекулярный путь). В то время как в реакции сочетания вновь образованное соединение алкил / арил диффундирует, и последующее окислительное присоединение происходит между входящей связью Ar-Br и металлическим центром. Ассоциативный комплекс необходим для того, чтобы полимеризация протекала живым образом, поскольку он позволяет металлу подвергаться предпочтительному внутримолекулярному окислительному присоединению и оставаться с единственной растущей цепью (в соответствии с механизмом роста цепи), в отличие от межмолекулярного окислительного присоединения другие мономеры, присутствующие в растворе (в соответствии со ступенчатым неживым механизмом роста).[24][25] Объем мономеров CTP увеличивался с момента его открытия и включал поли (фенилен) s, поли (фтор) s, поли (селенофен) s и поли (пиррол) s.[24][25]
Полимеризация с переносом живой группы
Полимеризация с переносом группы также имеет характеристики живой полимеризации.[26] Применяется для алкилированных метакрилат мономеры и инициатор силил кетен ацеталь. Новый мономер добавляется к инициатору и к активной цепи роста в Реакция Майкла. При каждом добавлении мономерной группы триметилсилильная группа переносится на конец цепи. Активный конец цепи не ионный, как при анионной или катионной полимеризации, но ковалентный. Реакция может катализироваться бифторидами и биоксианионами, такими как трис (диалкиламино) бифторид сульфония или же бибензоат тетрабутиламмония. Метод был открыт в 1983 г. О.В. Webster[27] и имя, впервые предложенное Барри Трост.
Приложения
Живые полимеризации используются в промышленном синтезе многих полимеров.
Синтез сополимеров и применение
Сополимеры представляют собой полимеры, состоящие из нескольких различных видов мономеров, и могут быть расположены в различном порядке, три из которых показаны на рисунке ниже.

Хотя существуют и другие (чередующиеся сополимеры, привитые сополимеры и стереоблок-сополимеры), эти три более распространены в научной литературе.[3] Кроме того, блок-сополимеры могут существовать многих типов, включая триблок (A-B-A), чередующийся блок (A-B-A-B-A-B) и т.д.
Из этих трех типов блок-сополимеры и градиентные сополимеры обычно синтезируются посредством живой полимеризации из-за простоты контроля живой полимеризации. Сополимеры очень желательны из-за повышенной гибкости свойств, которые полимер может иметь по сравнению с их гомополимерными аналогами. Используемые методы синтеза варьируются от ROMP до обычных анионных или катионных живых полимеризаций.[3][4]
Сополимеры, благодаря их уникальной возможности настройки свойств, могут иметь широкий спектр применения. Один из примеров (из многих) - наномасштаб. литография с использованием блок-сополимеров. Часто используется блок-сополимер из полистирола и полиметилметакрилата (сокращенно PS-б-ПММА). Этот сополимер при надлежащих термических условиях и условиях обработки может образовывать цилиндры из ПММА диаметром порядка нескольких десятков нанометров, окруженные матрицей из полистирола. Эти цилиндры затем можно протравить под воздействием ультрафиолетового излучения и уксусной кислоты, в результате чего останется пористая матрица из полистирола.[28][29][30]
Уникальное свойство этого материала заключается в том, что размер пор (или размер цилиндров из ПММА) можно легко регулировать с помощью соотношения ПС и ПММА при синтезе сополимера. Его можно легко настроить благодаря легкому контролю, обеспечиваемому реакциями живой полимеризации, что делает этот метод очень востребованным для создания различных наноразмерных рисунков из различных материалов для применения в катализе, электронике и т. Д.
Рекомендации
- ^ Халаса, А. Ф. (1981). «Последние достижения в анионной полимеризации». Химия и технология резины. 54 (3): 627–640. Дои:10.5254/1.3535823.
- ^ Моад, Грэм и Соломон, Дэвид Х. (2006) Химия радикальной полимеризации. 2-е изд. Эльзевир. ISBN 0-08-044286-2
- ^ а б c d е ж Cowie, J.M.G. (2007). Химия полимеров и физика современных материалов (3-е изд. / Изд. J.M.G. Cowie and Valeria Arrighi). Бока-Ратон: Тейлор и Фрэнсис. ISBN 9780849398131.
- ^ а б c d е ж грамм час я j k л Одиан, Джордж (2004). Принципы полимеризации (4-е изд.). Хобокен, Нью-Джерси: Wiley-Interscience. ISBN 978-0471274001.
- ^ Jenkins, A.D .; Kratochvíl, P .; Степто, Р. Ф. Т .; Сутер, У. В. (1996). «Глоссарий основных терминов в науке о полимерах (Рекомендации ИЮПАК 1996 г.)». Чистая и прикладная химия. 68 (12): 2287–2311. Дои:10.1351 / pac199668122287.
- ^ а б Szwarc, M .; Леви, М .; Милькович, Р. (1956). «Полимеризация, инициированная переносом электрона на мономер. Новый метод образования блочных полимеров1». Журнал Американского химического общества. 78 (11): 2656–2657. Дои:10.1021 / ja01592a101.
- ^ М. Шварц (1956). ""Живые «полимеры». Природа. 178 (4543): 1168. Дои:10.1038 / 1781168a0.
- ^ а б Макнил, Энн; Брайан, Захари (2013). «Доказательства предпочтительного внутримолекулярного окислительного присоединения в катализируемых Ni реакциях кросс-сочетания и их влияние на полимеризацию с ростом цепи». Chem. Наука. 4 (4): 1620–1624. Дои:10.1039 / C3SC00090G.
- ^ Шварц М. (1956). "'Живые «полимеры». Природа. 178 (4543): 1168–1169. Bibcode:1956Натура.178.1168S. Дои:10.1038 / 1781168a0.
- ^ Татемото, Масаёси и Накагава, Цунео «Сегментированные полимеры, содержащие фтор и йод, и их производство» Патент США 4,158,678 . Дата приоритета 30 июня 1976 г.
- ^ а б Craver, C .; Каррахер, К. (2000). Прикладная наука о полимерах: 21 век. Эльзевир. С. 1022–1023.
- ^ Коутс, Джеффри В. (апрель 2000 г.). «Точный контроль стереохимии полиолефинов с использованием одноцентровых металлических катализаторов». Химические обзоры. 100 (4): 1223–1252. Дои:10.1021 / cr990286u. PMID 11749265.
- ^ Jayaratne, K .; Сита, L (2000). «Стереоспецифическая живущая полимеризация Циглера-Натта 1-гексена». Варенье. Chem. Soc. 122 (5): 958–959. Дои:10.1021 / ja993808w.
- ^ Killian, C.M .; Tempel, D. J .; Johnson, L.K .; Брукхарт, М. (1996). «Живая полимеризация α-олефинов с использованием катализаторов NiII-α-диимина. Синтез новых блочных полимеров на основе α-олефинов». Журнал Американского химического общества. 118 (46): 11664–11665. Дои:10.1021 / ja962516h.
- ^ Brookhart, M .; Rix, F.C .; Desimone, J.M .; Барборак, Дж. К. (1992). «Катализаторы палладия (II) для живой чередующейся сополимеризации олефинов и окиси углерода». Журнал Американского химического общества. 114 (14): 5894–5895. Дои:10.1021 / ja00040a082.
- ^ а б Goethals, E; Дюпрез, Ф (2007). «Карбокатионная полимеризация». Прогресс в науке о полимерах. 32 (2): 220–246. Дои:10.1016 / j.progpolymsci.2007.01.001.
- ^ Schrock, R. R .; Feldman, J .; Канниццо, Л. Ф .; Граббс, Р. Х. (1987). «Полимеризация с раскрытием цикла норборнена живым комплексом алкилидена вольфрама». Макромолекулы. 20 (5): 1169–1172. Bibcode:1987MaMol..20.1169S. Дои:10.1021 / ma00171a053.
- ^ а б c Bielawski, Christopher W .; Граббс, Роберт Х. (2007). «Полимеризация метатезиса с раскрытием живого цикла». Прогресс в науке о полимерах. 32 (1): 1–29. Дои:10.1016 / j.progpolymsci.2006.08.006.
- ^ а б c Braunecker, Wade A .; Матияшевский, Кшиштоф (2007). «Управляемая / живая радикальная полимеризация: особенности, развитие и перспективы». Прогресс в науке о полимерах. 32 (1): 93–146. Дои:10.1016 / j.progpolymsci.2006.11.002.
- ^ Матяшевский. «Особенности контролируемой« живой »полимеризации». Архивировано из оригинал 14 марта 2014 г.
- ^ а б c Yokozawa, T .; Ёкояма, А. (2007). «Цепной рост поликонденсации: живой процесс полимеризации в поликонденсации». Прогресс в науке о полимерах. 32: 147–172. Дои:10.1016 / j.progpolymsci.2006.08.001.
- ^ а б Миякоши, Ре; Ёкояма, Акихиро; Ёкодзава, Цутому (2005). «Поликонденсация с переносом катализатора. Механизм катализируемой никелем полимеризации с цепным ростом, ведущей к четко определенному поли (3-гексилтиофену)». Журнал Американского химического общества. 127 (49): 17542–17547. Дои:10.1021 / ja0556880. PMID 16332106.
- ^ Йову, Михаэла Корина; Шеина, Елена Е .; Гил, Роберто Р .; Маккалоу, Ричард Д. (октябрь 2005 г.). «Экспериментальные доказательства квази-« живой »природы метода метатезиса Гриньяра для синтеза регулярных поли (3-алкилтиофенов)». Макромолекулы. 38 (21): 8649–8656. Bibcode:2005MaMol..38.8649I. CiteSeerX 10.1.1.206.3875. Дои:10.1021 / ma051122k.
- ^ а б Кирий, Антон; Сеньковский, Владимир; Зоммер, Майкл (4 октября 2011 г.). «Поликонденсация с переносом катализатора Кумада: механизм, возможности и проблемы». Макромолекулярные быстрые коммуникации. 32 (19): 1503–1517. Дои:10.1002 / marc.201100316. PMID 21800394.
- ^ а б Брайан, Захари Дж .; Макнил, Энн Дж. (12 ноября 2013 г.). «Синтез сопряженных полимеров с помощью поликонденсации с переносом катализатора (CTP): механизм, сфера применения и применение». Макромолекулы. 46 (21): 8395–8405. Bibcode:2013MaMol..46.8395B. Дои:10.1021 / ma401314x.
- ^ Дэвис, Фред Дж. (2004) Химия полимеров: практический подход. Издательство Оксфордского университета. ISBN 978-0-19-850309-5 .
- ^ Webster, O.W .; Hertler, W. R .; Sogah, D. Y .; Farnham, W. B .; Раджан Бабу, Т. В. (1983). «Полимеризация с переносом группы. 1. Новая концепция аддитивной полимеризации с кремнийорганическими инициаторами». Варенье. Chem. Soc. 105 (17): 5706–5708. Дои:10.1021 / ja00355a039.
- ^ In, Insik; Ла, Ён-Хе; Парк, Санг-Мин; Нили, Пол Ф .; Гопалан, Падма (август 2006 г.). "Щетки из случайных сополимеров с привитыми боковыми цепями как нейтральные поверхности для управления ориентацией микродоменов блочных сополимеров в тонких пленках". Langmuir. 22 (18): 7855–7860. Дои:10.1021 / la060748g. PMID 16922574.
- ^ Лю, Юаньцзюнь; Гонг, Яньчунь; Он, Лонгбин; Се, Бо; Чен, Си; Хан, Мин; Ван, Гуангоу (2010). «Формирование периодических массивов наноколец на самоорганизующейся пленке ПС-b-ПММА при быстром отжиге в растворителе». Наномасштаб. 2 (10): 2065–8. Bibcode:2010Nanos ... 2.2065L. Дои:10.1039 / c0nr00207k. PMID 20820641.
- ^ Эдвардс, Э. У .; Монтегю, М. Ф .; Solak, H.H .; Hawker, C.J .; Нили, П. Ф. (4 августа 2004 г.). «Точный контроль над молекулярными размерами доменов блок-сополимеров с использованием межфазной энергии субстратов с химическим наноразмером». Современные материалы. 16 (15): 1315–1319. Дои:10.1002 / adma.200400763.