Стилле реакция - Stille reaction
| Стилле реакция | |
|---|---|
| Названный в честь | Джон Кеннет Стилл |
| Тип реакции | Реакция сцепления |
| Идентификаторы | |
| Портал органической химии | ходовая часть |
| RSC ID онтологии | RXNO: 0000035 |
В Стилле реакция это химическая реакция широко используется в органический синтез. Реакция включает связывание двух органических групп, одна из которых переносится как оловоорганическое соединение (также известный как органостаннаны). Разнообразные органические электрофилы обеспечивают другое партнер по связке. Реакция Стилла - одна из многих реакции сочетания, катализируемые палладием.[1][2][3]
- : Аллил, алкенил, арил, бензил, ацил.
- : галогениды (Cl, Br, I), псевдогалогениды (OTf, ), OAc
![{ displaystyle { color {Blue} { ce {R ^ {1} -Sn (Alkyl) 3}}} + { color {Red} { ce {R ^ {2} -X}}} { ce {-> [{ color {Green} { ce {Pd ^ {0}}}} { text {(каталитический)}}] [{ text {набор лигандов}}]}} overbrace { { color {Blue} { ce {R ^ {1}}}} ! - ! { color {Red} { ce {R ^ {2}}}}} ^ {connected product} + { color {Red} { ce {X}}} ! - ! { color {Blue} { ce {Sn (алкил) 3}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/5baabb66db61c2d31fa2a5ca2b4e8156ee7c4133 "Общая схема реакции Стилле")



R1 группа, присоединенная к триалкилолову, обычно sp2-гибридизированные, в том числе винил, и арил группы.
Эти органостаннаны также устойчивы как к воздуху, так и к влаге, и многие из этих реагентов либо коммерчески доступны, либо могут быть синтезированы на основании прецедентов в литературе. Однако эти реагенты с оловом очень токсичны. X обычно галогенид, Такие как Cl, Br, или же я, но псевдогалогениды, такие как трифлаты и сульфонаты и фосфаты также можно использовать.[4][5] Опубликовано несколько обзоров.[6][2][7][8][9][10][11][12][13][14][15]
История
Первый пример катализируемое палладием сочетание арилгалогенидов с оловоорганические реагенты сообщил Колин Иборн в 1976 г.[16] Эта реакция дает от 7% до 53% диарильного продукта. Этот процесс был расширен до объединения ацилхлориды с реагентами алкил-олова в 1977 г. компанией Toshihiko Migita, выход от 53% до 87% кетон товар.[17]

В 1977 году Migita опубликовала дальнейшую работу по объединению аллил -оловянные реагенты с обоими арил (C) и ацил (D) галогениды. Большая способность аллильных групп мигрировать в палладиевый катализатор позволял проводить реакции при более низких температурах. Выходы арилгалогенидов составляют от 4% до 100%, а ацилгалогенидов - от 27% до 86%.[18][19] Отражая ранний вклад Мигиты и Косуги, реакцию Стилле иногда называют Муфта Мигита – Косуги – Стилле.

Stille впоследствии сообщили о сочетании различных реагентов на основе алкилолова в 1978 году с многочисленными арил- и ацилгалогенидами в мягких условиях реакции с гораздо лучшими выходами (76% -99%).[18][20] В 1980-х годах Стилле продолжил свою работу по синтезу множества кетонов, используя этот широкий и мягкий процесс, и выяснил механизм этого превращения.[21][22]

К середине 1980-х было опубликовано более 65 статей на тему реакций сочетания с участием олова, в которых продолжалось изучение субстратных возможностей этой реакции. В то время как первоначальные исследования в этой области были сосредоточены на связывании алкильных групп, большая часть будущих работ включала гораздо более синтетически полезное соединение винил, алкенил, арил и аллил органостаннаны к галогенидам. Из-за устойчивости этих оловоорганических реагентов к воздуху и легкости их синтеза реакция Стилле стала обычным явлением в органическом синтезе.[8]
Механизм
Механизм реакции Стилле широко изучен.[11][23] В каталитический цикл включает в себя окислительная добавка из галогенид или псевдогалогенид (2) к палладиевый катализатор (1), трансметалляция из 3 с оловоорганический реагент (4), и восстановительное устранение из 5 чтобы получить связанный продукт (7) и регенерированный палладиевый катализатор (1).[24]

Однако детальный механизм связывания Стилле чрезвычайно сложен и может происходить через многочисленные реакционные пути. Как и другие реакции сочетания, катализируемые палладием активный палладиевый катализатор считается 14-электронным комплексом Pd (0), который может быть образован различными способами. Использование 18- или 16-электронного источника Pd (0) Pd (PPh3)4, Pd (дБа)2 может пройти лиганд диссоциация с образованием активных разновидностей. Второй, фосфины может быть добавлен к безлигандному палладию (0). Наконец, как на фото, снижение источника Pd (II) (8) (Pd (OAc)2, PdCl2(MeCN)2, PdCl2(PPh3)2, BnPdCl (PPh3)2и т. д.) путем добавления фосфиновых лигандов или оловоорганических реагентов. [6]
Окислительное добавление
Предложено окислительное присоединение к 14-электронному комплексу Pd (0). Этот процесс дает 16-электронное соединение Pd (II). Было высказано предположение, что анионные лиганды, Такие как OAc, ускорить этот шаг за счет образования [Pd (OAc) (PR3)п]−, что делает частицы палладия более нуклеофильными.[11][25]В некоторых случаях, особенно когда зр3-гибридизированный органо-галогенид используется, SN2-х типовой механизм имеет тенденцию преобладать, но это не так часто встречается в литературе.[11][25]Однако, несмотря на нормальное формирование СНГ-сразу после согласованное окислительное добавление, этот товар быстро равновесие с этими транс-изомер.[26][27]

Трансметалляция
В трансметалляция из транс промежуточный от окислительная добавка Считается, что стадия протекает посредством множества механизмов в зависимости от субстратов и условий. Наиболее распространенный тип трансметалляции муфты Стилле включает ассоциативный механизм. Этот путь подразумевает, что органостаннан, обычно банка атом, связанный с аллильной, алкенильной или арильной группой, может координировать к палладию через одну из этих двойных связей. Это дает мимолетный пятивалент, 18-электронные разновидности, который затем может подвергнуться отщеплению лиганда с образованием квадратный плоский снова комплекс. Несмотря на то, что органостаннан координируется с палладием через R2 группа, R2 должны быть официально переданы палладий (R2-Sn связь должна быть разорвана), и группа X должна уйти вместе с оловом, завершая трансметалляцию. Считается, что это происходит за счет двух механизмов.[28]
Во-первых, когда органостаннан первоначально присоединяется к транс-металлическому комплексу, группа X может координировать к банка, помимо палладия, образуя циклический переходное состояние. Разрушение этого аддукта приводит к потере R3Sn-X и трехвалентный палладий комплекс с R1 и R2 присутствует в СНГ отношение. Другой часто встречающийся механизм включает такое же первоначальное добавление станнана к транс комплекс палладия, как показано выше; однако в этом случае группа X не координируется с оловом, создавая открытый переходное состояние. После α-углерод по сравнению с оловом, атакующим палладий, комплекс олова уйдет с чистым положительным зарядом. На схеме ниже обратите внимание, что двойная связь, координирующая олово, обозначает R2так что любой алкенил, аллил, или же арил группа. Кроме того, группа X может диссоциировать в любое время в течение механизма и связываться с Sn+ сложный в конце. Функциональная теория плотности расчеты предсказывают, что открытый механизм будет преобладать, если 2 лиганды остаются прикрепленными к палладию, и группа X уходит, в то время как циклический механизм более вероятен, если лиганд диссоциирует до трансметалляция. Следовательно, хорошие уходящие группы, такие как трифлаты в полярных растворителях, благоприятствуют первым, в то время как объемные фосфиновые лиганды будет отдавать предпочтение последнему.[28]

Менее распространенный путь для трансметалляция проходит через диссоциативный или с помощью растворителя механизм. Здесь лиганд четырехвалентного палладия диссоциирует, и координирующий растворитель может добавляться к палладию. Когда растворитель отделяется с образованием трехвалентного интермедиата с 14 электронами, органостаннан можно добавить к палладий, подвергаясь процессу открытого или циклического типа, как указано выше.[28]
Восстановительный этап устранения
Для того, чтобы R1-Р2 к редуктивно устранить, эти группы должны занимать взаимно СНГ координационные сайты. Любой транс-аддукты должны поэтому изомеризоваться в СНГ промежуточный, иначе муфта будет нарушена. Существует множество механизмов восстановительного устранения, и они обычно считаются согласованными.[11][29][30]
Во-первых, 16-электронная четырехвалентный промежуточный от трансметалляция шаг может подвергнуться восстановительному исключению без посторонней помощи из квадратный плоский сложный. Эта реакция происходит в два этапа: во-первых, за восстановительным элиминированием следует координация вновь образованной сигма-связи между R1 и R2 к металлу, с окончательной диссоциацией с образованием связанного продукта.[11][29][30]

Однако предыдущий процесс иногда бывает медленным и может быть значительно ускорен диссоциацией лиганда с образованием 14-электронного Т-образный промежуточный. Этот промежуточный продукт может затем перегруппироваться с образованием Y-образного аддукта, который может подвергаться более быстрому восстановительному отщеплению.[11][29][30]

Наконец, дополнительный лиганд может ассоциироваться с палладием с образованием 18-электронной тригональной бипирамидальной структуры с R1 и R2 цис друг к другу в экваториальных положениях. Геометрия этого промежуточного звена делает его похожим на Y-образную форму выше.[11][29][30]

Наличие объемные лиганды также может увеличить скорость выведения. Лиганды, такие как фосфины с большим углы прикуса причина стерическое отталкивание между L и R1 и R2, в результате чего угол между L и R группами увеличивается, а угол между R1 и R2 чтобы, следовательно, уменьшить, что позволяет быстрее восстановительное устранение.[11][24]

Кинетика
Скорость, с которой органостаннаны трансметаллировать с палладиевыми катализаторами показано ниже. Sp2-гибридизированные углеродные группы, присоединенные к олову, являются наиболее часто используемыми партнерами связывания, а sp3-гибридизированные атомы углерода требуют более жестких условий, и концевые алкины могут быть связаны через связь C-H через Соногашира реакция.

В качестве органического соединения олова обычно используют триметилстаннильное или трибутилстаннильное соединение. Хотя триметилстаннильные соединения демонстрируют более высокую реакционную способность по сравнению с трибутилстаннильными соединениями и имеют гораздо более простые 1Спектры H-ЯМР, токсичность первого намного выше.[31]
Оптимизация того, какие лиганды лучше всего подходят для проведения реакции с высоким выходом и скоростью оборота, может быть трудной. Это потому, что окислительная добавка требуется металл, богатый электронами, следовательно, предпочтение отдается электронодонорным лигандам. Однако металл с дефицитом электронов более благоприятен для трансметалляция и восстановительное устранение шаги, делая электроноакцепторные лиганды здесь лучшими. Таким образом, оптимальный набор лигандов сильно зависит от индивидуальных используемых субстратов и условий. Они могут изменить шаг определения скорости, а также механизм трансметалляция шаг.[32]
Обычно используются лиганды с промежуточной донностью, такие как фосфины. Повышение скорости можно увидеть при использовании лигандов с умеренным содержанием электронов, таких как три-2-фурилфосфин или трифениларсенин. Точно так же лиганды с большим числом доноров могут замедлять или ингибировать реакции связывания.[32][33]
Эти наблюдения предполагают, что обычно этап определения скорости реакции Стилле трансметалляция.[33]
Добавки
Самая распространенная добавка к реакции Стилле - это стехиометрический или же сокаталитический медь (I), конкретно йодид меди, что может улучшить тарифы больше на> 103 складывать. Было высказано предположение, что в полярных растворители медь трансметаллировать с органостаннан. Результирующий органокупрат реагент может затем трансметаллировать с палладиевым катализатором. Кроме того, в эфирных растворителях медь также может способствовать удалению фосфиновый лиганд, активируя центр Pd.[9][34][35][36][37]
Лития хлорид было обнаружено, что он является мощным ускорителем скорости в случаях, когда группа X диссоциирует от палладия (т. е. открытый механизм). В хлористый ион, как полагают, либо замещает группу X на палладии, делая катализатор более активным для трансметалляция или путем координации с аддуктом Pd (0) для ускорения окислительная добавка. Кроме того, соль LiCl усиливает полярность растворителя, облегчая этот обычно анионный лиганд (–Cl, –Br, –OTf и т. д.) оставить. Эта добавка необходима, когда такие растворители, как THF используется; однако использование более полярного растворителя, такого как NMP, может заменить потребность в этой солевой добавке. Однако, когда стадия трансметаллирования муфты протекает по циклическому механизму, добавление хлорида лития может фактически снизить скорость. Как и в циклическом механизме, нейтральный лиганд, такой как фосфин, должен диссоциировать вместо анионной группы X.[10][38]
Наконец, источники ионы фтора, Такие как фторид цезия, также влияют на каталитический цикл. Во-первых, фторид может увеличивать скорость реакции органотрифлаты, возможно, тем же эффектом, что и хлорид лития. Кроме того, ионы фтора могут действовать как мусорщики за банка побочные продукты, что упрощает их удаление через фильтрация.[36]
Конкурирующие побочные реакции
Наиболее распространенная побочная реакционная способность, связанная с реакцией Стилле, - это гомосочетание реагентов станнана с образованием R2-Р2 димер. Считается, что это происходит с помощью двух возможных механизмов. Во-первых, реакция двух эквивалентов органостаннан с предкатализатором Pd (II) будет давать гомо-связанный продукт после восстановительное устранение. Во-вторых, катализатор Pd (0) может подвергаться радикальный процесс чтобы получить димер. Используемый органостаннановый реагент традиционно четырехвалентен по олову, обычно состоящий из пр.2-гибридизованная группа для переноса и три «непередаваемых» алкил группы. Как видно выше, алкильные группы обычно наиболее медленно мигрируют на палладиевый катализатор.[10]

Также было обнаружено, что при температуре до 50 ° C, арил группы на обоих палладий и скоординированный фосфин можно обменять. Хотя обычно они не обнаруживаются, во многих случаях они могут быть второстепенным продуктом.[10]
Наконец, довольно редкая и экзотическая побочная реакция известна как подмена кино. Здесь после начального окислительная добавка из арилгалогенид, эта разновидность Pd-Ar может вставляться через двойную связь винил-олова. После устранение β-гидрида, миграционная вставка, и протодестаннилирование, можно синтезировать 1,2-дизамещенный олефин.[10]

Могут возникнуть многочисленные другие побочные реакции, в том числе: E / Z изомеризация, что потенциально может быть проблемой при использовании алкенилстаннана. Механизм этого превращения в настоящее время неизвестен. Обычно, органостаннаны довольно устойчивы к гидролиз, тем не менее, когда используются арилстаннаны с очень высоким содержанием электронов, это может стать значительной побочной реакцией.[10]
Объем
Электрофил
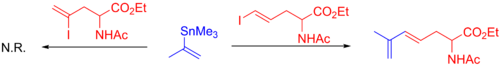
Галогениды винила являются общими партнерами в реакции Стилле, и реакции этого типа встречаются во многих натуральный продукт общий синтез. Обычно используются винилйодиды и бромиды. Винилхлориды недостаточно реактивны по отношению к окислительная добавка в Pd (0). Йодиды обычно предпочтительны: они обычно реагируют быстрее и в более мягких условиях, чем бромиды. Это различие демонстрируется ниже выборочным связь винилйодида в присутствии винилбромида.[10]
Обычно стереохимия из алкен сохраняется на протяжении всей реакции, за исключением жестких условий реакции. Могут использоваться различные алкены, и они включают как α-, так и β-галоген-α, β-ненасыщенные кетоны, сложные эфиры, и сульфоксиды (для чего обычно требуется добавка меди (I)) и многое другое (см. пример ниже).[39] Иногда используются также винилтрифлаты. Некоторые реакции требуют добавления LiCl а другие замедляются, что подразумевает наличие двух механистических путей.[10]

Другой класс обычных электрофилы арил и гетероциклический галогениды. Что касается виниловых подложек, бромиды и йодиды встречаются чаще, несмотря на их большую стоимость. Можно выбрать множество арильных групп, включая кольца, замещенные электронодонорными заместителями, биарил кольца и многое другое. Галогензамещенный гетероциклы также использовались в качестве партнеров по сцеплению, в том числе пиридины, фураны, тиофены, тиазолы, индолы, имидазолы, пурины, урацил, цитозины, пиримидины и многое другое (см. ниже таблицу гетероциклов; галогены могут быть замещены во множестве положений на каждом).[10]

Ниже приведен пример использования сцепления Stille для создания сложности на гетероциклы из нуклеозиды, Такие как пурины.[40]

Арил трифлаты и сульфонаты также сочетаются с широким спектром станнановых реагентов. Трифлаты имеют тенденцию реагировать сравнимо с бромидами в реакции Стилле.[10]
Ацилхлориды также используются в качестве партнеров для связывания и могут использоваться с широким спектром станнана, даже с реагентами алкил-олова, для получения кетоны (см. пример ниже).[41] Однако иногда бывает трудно ввести ацилхлорид. функциональные группы на большие молекулы с чувствительными функциональными группами. Альтернативой, разработанной для этого процесса, является реакция перекрестного связывания с карбонилированием Стилле, которая вводит карбонил группа через введение окиси углерода.[10]

Аллильный, бензиловый, и пропаргиловый галогениды также могут быть связаны. Обычно используемые аллильные галогениды протекают через η3 переходное состояние, позволяющее связываться с органостаннаном в α- или γ-положении, преимущественно по наименее замещенному углероду (см. пример ниже).[42] Алкениловые эпоксиды (смежные эпоксиды и алкены ) также может претерпевать такую же связь через η3 переходное состояние as, открывая эпоксид алкоголь. В то время как аллильный и бензиловый ацетаты Обычно используются пропаргиловые ацетаты, которые не реагируют с органостаннанами.[10]

Станнан
Органостаннановые реагенты общие. Некоторые из них имеются в продаже.[43] Станнановые реагенты могут быть синтезированы по реакции Гриньяр или же литийорганический реагент с хлоридами триалкилолова. Например, винилтрибутилолово получают реакцией бромида винилмагния с хлоридом трибутилолова.[44] Гидростаннилирование из алкины или же алкены предоставляет множество производных. Оловоорганические реагенты устойчивы к воздуху и влаге. Некоторые реакции могут происходить даже в воде.[45] Их можно очистить хроматография. Они толерантны к большинству функциональных групп. Некоторые оловоорганические соединения сильно токсичный, особенно производные триметилстаннила.[10]
Широко распространено использование реагентов на основе винилстаннана или алкенилстаннана.[10] Что касается ограничений, как очень объемные реагенты станнана, так и станнаны с заменой на α-углерод как правило, вяло реагируют или требуют оптимизации. Например, в приведенном ниже случае α-замещенный винилстаннан реагирует только с концевым иодидом из-за стерическое препятствие.[46]
Реагенты арилстаннана также широко распространены, и оба пожертвование электронов и вывод электронов группы фактически увеличивают скорость трансметалляции. Это снова означает, что два механизма трансметалляция может случиться. Единственным ограничением для этих реагентов являются заместители в орто-положении, настолько малые, что метильные группы могут снизить скорость реакции. Большое разнообразие гетероциклы (см. раздел «Электрофилы») также могут использоваться в качестве партнеров по связи (см. пример с тиазол кольцо внизу).[10][47]


Алкинилстаннаны, наиболее реактивные из станнанов, также использовались в муфтах Стилле. Обычно они не нужны, поскольку концевые алкины могут напрямую связываться с палладиевыми катализаторами через их связь C-H через Муфта Соногашира. Сообщалось, что аллилстаннаны работают, однако возникают трудности, как и с аллильными галогенидами, с трудностью контроля региоселективность для сложения α и γ. Реагенты дистаннан и ацилстаннан также использовались в муфтах Стилле.[10]
Приложения
Реакция Стилле использовалась в синтезе множества полимеров.[48][49][50] Однако наиболее распространенным использованием реакции Стилле является ее использование в органический синтез, а именно в синтезе натуральные продукты.
Полный синтез натурального продукта
Овермана 19 ступеней энантиоселективный полный синтез квадригема С включает двойной стиль перекрестный метатезис реакция.[6][51] Комплексный станнан присоединен к двум арилиодидным группам. После двойного Черт циклизация, продукт достигается.

32 ступень Панека энантиоселективный полный синтез из ансамицин антибиотик (+) - микотриенол использует тандемное соединение макроцикла типа Стилле на поздней стадии. Здесь органостаннан имеет две концевые группы трибутилолова, атакованные до алкена. Этот органостаннан «сшивает» два конца линейного исходного материала в макроцикл, добавляя в процесс два недостающих метиленовых звена. После окисления ароматической сердцевины аммиачная селитра церия (CAN) и снятие защиты с плавиковая кислота дает натуральный продукт с выходом 54% за 3 стадии.[6][52]

Стивен Ф. Мартин а в 21-ступенчатом энантиоселективном полном синтезе манзамина противоопухолевого алкалоида Ircinal A коллег используется тандемная реакция Стилла / Дильса-Альдера в одном сосуде. К винилбромиду добавляется алкеновая группа, за которой следует на месте Дильс-Альдер циклоприсоединение между добавленным алкеном и алкеном в пирролидин звенеть.[6][53]

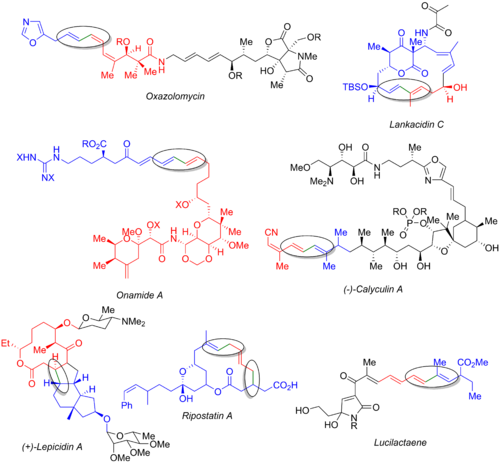
Во многих других полных синтезах используется реакция Стилле, включая оксазоломицин,[54] ланкацидин С,[55] онамид А,[56] каликулин А,[57] лепицидин А,[58] рипостатин А,[59] и люцилактаен.[6][60] На изображении ниже показан последний натуральный продукт, галогенид (синий), станнанорганический (красный) и образующаяся связь (зеленый и обведен). Из этих примеров ясно, что реакция Стилле может использоваться как на ранних стадиях синтеза (оксазоломицин и каликулин A), так и в конце конвергентного пути (онамид A, ланкацидин C, рипостатин A) или в средний (лепицидин А и люцилактаен). Синтез рипостатина А включает два одновременных связывания Стилле, за которыми следует метатезис замыкающего кольца. Синтез луцилактаена включает среднюю субъединицу, имеющую боран с одной стороны и станнан с другой, что позволяет проводить реакцию Стилле с последующим сочетанием Сузуки.
Вариации
Помимо проведения реакции в различных органических растворителях, были разработаны условия, которые позволяют использовать широкий диапазон сочетаний Стилле в водном растворителе.[14]
В присутствии солей Cu (I) палладий на угле было показано, что это эффективный катализатор.[61][62]
В сфере зеленая химия Сообщается, что реакция Стилле происходит в легкоплавкой и высокополярной смеси сахара, такой как маннитол, а мочевина такие как диметилмочевина и соль, такая как хлорид аммония[63].[64] Каталитическая система трис (дибензилиденацетон) дипалладий (0) с трифениларсин:

Стилле-карбонилированное кросс-сочетание
Распространенным изменением муфты Stille является включение карбонил группа между R1 и R2, служащий эффективным методом для формирования кетоны. Этот процесс очень похож на первоначальное исследование Migita и Stille (см. Историю) сочетания станнана с ацилхлориды. Однако эти фрагменты не всегда легко доступны, и их может быть трудно сформировать, особенно в присутствии чувствительных функциональные группы. Кроме того, может быть сложно контролировать их высокую реактивность. Соединение Стилле-карбонилирование происходит в тех же условиях, что и соединение Стилле, за исключением того, что в атмосфере монооксид углерода (CO) используется. CO может координироваться с палладиевым катализатором (9) после первоначального окислительного добавления с последующим Вставка CO в Pd-R1 связь (10), в результате чего восстановительное устранение к кетону (12). В трансметалляция шаг обычно этап определения ставки.[6]

Ларри Оверман и коллеги используют перекрестное связывание Стилле-карбонилирование в своих 20-этапных энантиоселективный полный синтез из стрихнин. Добавленный карбонил позже превращается в концевой алкен через Реакция Виттига, что позволяет формировать ключевой третичный азот и пентациклическое ядро через аза-Справиться -Реакция Манниха.[6][65]
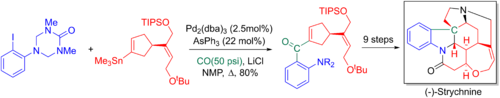
Джорджио Ортар и др. исследовали, как перекрестное связывание с карбонилированием Стилле может быть использовано для синтеза бензофенон фосфоры. Они были включены в 4-бензоил-L-фенилаланин. пептиды и использовались из-за их фотоаффинных свойств мечения для изучения различных пептидно-белковых взаимодействий.[6][66]

16 шагов Луи Хегедуса рацемический полный синтез джатрафона включает в себя перекрестное связывание с карбонилированием Стилла в качестве заключительной стадии образования 11-членного макроцикл. Вместо града в качестве связующего звена используется винилтрифлат.[6][67]

Муфта Стилла – Келли
Используя основополагающую публикацию Eaborn в 1976 г., который образует арилстаннаны из арилгалогенидов и дистаннанов, Т. Росс Келли применил этот процесс к внутримолекулярный сочетание арилгалогенидов. Это тандемное сочетание станнилирование / арилгалогенид использовали для синтеза различных дигидрофенантренов. Большинство образующихся внутренних колец ограничено 5 или 6 членами, однако сообщалось о некоторых случаях макроциклизации. В отличие от обычного соединения Стилле, хлор не работает как галоген, возможно, из-за его более низкой реакционной способности в галоген последовательность (более короткая длина связи и более сильная энергия диссоциации связи затрудняют разрыв через окислительная добавка ). Начиная с середины схемы ниже и двигаясь по часовой стрелке, палладиевый катализатор (1) окислительно добавляет к наиболее реактивной связи C-X (13) сформировать 14, с последующим трансметалляция с дистананнане (15) уступить 16 и восстановительное устранение чтобы получить арилстаннан (18). Регенерированный палладиевый катализатор (1) может окислительная добавка ко второй связи C-X 18 формировать 19с последующим внутримолекулярным трансметалляция уступить 20, с последующим восстановительное устранение чтобы получить связанный продукт (22).[6]

Джи Джек Ли и др. использовали сочетание Стилла-Келли в своем синтезе различных кольцевых систем бензо [4,5] фуропиридинов. Они вызывают трехэтапный процесс, включающий Аминирование Бухвальда-Хартвига, еще один реакция сочетания, катализируемая палладием с последующим внутримолекулярным взаимодействием Стилла-Келли. Обратите внимание, что арил-йодидная связь будет окислительно добавить к палладий быстрее, чем любая из арилбромидных связей.[6][68]
![Синтез бензо [4,5] фуропиридинов.](http://upload.wikimedia.org/wikipedia/commons/thumb/2/25/Benzofuropyridines.png/500px-Benzofuropyridines.png)
Смотрите также
- Оловоорганическая химия
- Добавка органостаннана
- Катализируемые палладием реакции сочетания
- Сузуки реакция
- Муфта Негиши
- Чертовски реакция
- Муфта Хияма
Рекомендации
- ^ Хартвиг, Дж. Ф. Химия органических переходных металлов, от связывания до катализа; Научные книги университета: Нью-Йорк, 2010. ISBN 189138953X
- ^ а б Стилле, Дж. К. Энгью. Chem. Int. Эд. Англ. 1986, 25, 508–524. (Рассмотрение )
- ^ Фарина, В .; Кришнамурти, V .; Скотт, В. Дж. Орг. Реагировать. 1998, 50, 1–652. (Рассмотрение )
- ^ Scott, W. J .; Crisp, G.T .; Стилле, Дж. К. Органический синтез, Сб. Vol. 8, стр. 97 (1993); Vol. 68, стр. 116 (1990). (Статья )
- ^ Stille, J. K .; Эчаваррен, А. М .; Williams, R.M .; Хендрикс, Дж. А. Органический синтез, Сб. Vol. 9, с. 553 (1998); Vol. 71, стр.97 (1993). (Статья )
- ^ а б c d е ж грамм час я j k л Kurti, L .; Чако, Б. Стратегические применения названных реакций в органическом синтезе; Эльзевир: Берлингтон, 2005.
- ^ Митчелл, Т. J. Organomet. Chem., 1986, 304, 1-16.
- ^ а б Митчелл, Т. Синтез, 1992, 803-815. (Дои:10.1055 / с-1992-26230 )
- ^ а б Фарина, В. Pure Appl. Chem., 1996, 68, 73–78. (Дои:10.1351 / pac199668010073 ).
- ^ а б c d е ж грамм час я j k л м п о п Фарина, В .; Кришнамурти, V .; Скотт, В. Дж. Реакция Стиля; Wiley: Online, 2004. (Дои:10.1002 / 0471264180.or050.01 ).
- ^ а б c d е ж грамм час я Espinet, P .; Эчаваррен, А.М. Энгью. Chem. Int. Эд., 2004, 43, 4704–4734.(Дои:10.1002 / anie.200300638 )
- ^ Pattenden, G .; Синклер, Д. Дж. J.Organomet. Chem., 2002, 653, 261-268.
- ^ Kosugi, M .; Фугами, К. J. Organomet. Chem., 2002, 19, 10-16.
- ^ а б Pierre Genet, J .; Савиньяк, М. J. Organomet. Chem., 1999, 576, 305-317.
- ^ Кордова, С .; Bartolomé, C .; Martínez-Ilarduya, J.M ..; Эспине, П. ACS Catal., 2015, 5, 3040–3053.(Дои:10.1021 / acscatal.5b00448 ).
- ^ Азарян, Д .; Dua, S. S .; Eaborn, C .; Уолтон, Д. Р. М. J. Organomet. Chem., 1976, 117, C55-C57. (Дои:10.1016 / S0022-328X (00) 91902-8 )
- ^ Kosugi, M .; Shimizu, Y .; Мигита, Т. Chem. Lett., 1977, 6, 1423-1424. (Дои:10.1246 / cl.1977.1423 )
- ^ а б Kosugi, M .; Sasazawa, K .; Shikizu, Y .; Мигита, Т. Chem. Lett., 1977, 6, 301-302. (Дои:10.1246 / класс.1977.301 )
- ^ Kosugi, M .; Shimizu, Y .; Мигита, Т. J. Organomet. Chem., 1977, 129, C36-C38. (Дои:10.1016 / S0022-328X (00) 92505-1 )
- ^ Milstein, D .; Стилле, Дж. К. Варенье. Chem. Soc., 1978, 100, 3636-3638. (Дои:10.1021 / ja00479a077 )
- ^ Milstein, D .; Стилле, Дж. К. Варенье. Chem. Soc., 1979, 101, 4992-4998. (Дои:10.1021 / ja00511a032 )
- ^ Milstein, D .; Стилле, Дж. К. J. Org. Chem., 1979, 44, 1613-1618. (Дои:10.1021 / jo01324a006 )
- ^ Casado, A. L .; Espinet, P .; Гальего, А.М. J. Am, Chem. Soc., 2000, 122, 11771-11782. (Дои:10.1021 / ja001511o )
- ^ а б Крэбтри, Р. Х. Металлоорганическая химия переходных металлов., 5 изд .; Wiley: Нью-Йорк, 2009.
- ^ а б Perez-Temprano, M. H .; Гальего, А. М .; Casares, J. A .; Эспине, П. Металлоорганические соединения, 2011, 30, 611-617. (Дои:10.1021 / om100978w ).
- ^ Миннити, Д. Неорг. Chem, 1994, 33, 2631-2634.(Дои:10.1021 / ic00090a025 ).
- ^ Casado, A. L .; Эспине, П. Металлоорганические соединения, 1998, 17, 954-959. (Дои:10.1021 / om9709502 ).
- ^ а б c Гарсия-Мельчор, М .; Braga, A.A.C .; Lledos, A .; Ujaque, G .; Мазерас, Ф. Соотв. Chem. Res., 2013, 46, 2626-2634. (Дои:10.1021 / ar400080r )
- ^ а б c d Гилли, А .; Стилле, Дж. К. Варенье. Chem. Soc., 1980, 102, 4933-4941. (Дои:10.1021 / ja00535a018 ).
- ^ а б c d Brown, J.M .; Кули, Н.А. Chem. Ред., 1988, 88, 1031-1046. (Дои:10.1021 / cr00089a003 ).
- ^ McKillop, A .; Abel, E.W .; Stone, F.G.A .; Уилкинсон, Г. Комплексная металлоорганическая химия II, Elsevier Scientific: Оксфорд, 1995.
- ^ а б Фарина, В .; Варенье. Chem. Soc., 1991, 113, 9585-9595. (Дои:10.1021 / ja00025a025 ).
- ^ а б http://hwpi.harvard.edu/files/myers/files/11-the_stille_reaction.pdf
- ^ Либескинд, Л. С .; Фенгл, Р.В. J. Org. Chem., 1990, 55, 5359-5364. (Дои:10.1021 / jo00306a012 ).
- ^ Фарина, В .; Kapadia, S .; Brishnan, B .; Wang, C .; Либескинд, Л.С. J, Org. Chem, 1994, 59, 5905-5911. (Дои:10.1021 / jo00099a018 ).
- ^ а б Mee, S.P.H .; Ли, В .; Болдуин, Дж. Э. Энгью. Chem. Int. Эд., 2004, 43, 1132-1136.
- ^ Либескинд, Л. С .; Пенья-Кабрера, Э. Органический синтез, Сб. Vol. 10, стр.9 (2004); Vol. 77, с. 135 (2000). (Статья )
- ^ Scott, W. J .; Стилле, Дж. К. Варенье. Chem. Soc., 1986, 108, 3033-3040. (Дои:10.1021 / ja00271a037 ).
- ^ Johnson, C. R .; Adams, J. P .; Braun, M.P .; Сенанаяке, К. Б. У. Tetrahedron Lett., 1992, 33, 919-922. (Дои:10.1016 / S0040-4039 (00) 91576-4 )
- ^ Наир, В .; Тернер, Г. А .; Чемберлен, С. Варенье. Chem. Soc., 1987, 109, 7223-7224. (Дои:10.1021 / ja00257a071 ).
- ^ Jousseaume, B .; Kwon, W .; Verlhac, J. B .; Denat, F .; Дубац, Дж. Synlett, 1993, 117-118. (Дои:10.1055 / с-1993-22368 )
- ^ Шеффи, Ф. К .; Godschalx, J. P .; Стилле, Дж. К. Варенье. Chem. Soc., 1984, 106, 4833-4840. (Дои:10.1021 / ja00329a032 )
- ^ http://www.sigmaaldrich.com/chemistry/chemistry-products.html?TablePage=16246425
- ^ Дитмар Сейферт (1959). "Ди-п-бутилдивинилолово ". Орг. Синтезатор. 39: 10. Дои:10.15227 / orgsyn.039.0010.
- ^ Wolf, C .; Леребур, Р. J. Org. Chem., 2003,68 7551-7554. (Дои:10.1021 / jo0347056 ).
- ^ Crisp, G.T .; Глинк, П.Т. Тетраэдр, 1994, 50, 2623. (Дои:10.1016 / S0040-4020 (01) 86978-7 )
- ^ Бейли, Т. Tetrahedron Lett., 1986, 27, 4407. (Дои:10.1016 / S0040-4039 (00) 84964-3 ).
- ^ Bao, Z .; Чан, Вт .; Ю, Л. Chem. Матер., 1993, 5, 2-3. (Дои:10.1021 / см 00025a001 ).
- ^ Bao, Z .; Chan, W. K .; Ю., Л. Варенье. Chem. Soc., 1995, 117, 12426-12435. (Дои:10.1021 / ja00155a007 ).
- ^ Sun, S. S .; Lewis, J. E .; Zhang, J .; Цзян, X .; Zhang, C .; Matos, T .; Li, R .; Polym. Chem., 2010, 1, 663-669. (Дои:10.1039 / B9PY00324J )
- ^ Lebsack, A.D .; Link, J. T .; Overman, L.E .; Стернс, Б.А. Варенье. Chem. Soc., 2002, 124, 9008-9009. (Дои:10.1021 / ja0267425 )
- ^ Masse, C.E .; Ян, М .; Соломон, Дж .; Панек, Дж. С. Варенье. Chem. Soc., 1998, 120, 4123-4134. (Дои:10.1021 / ja9743194 )
- ^ Мартин, С. Ф .; Хамфри, Дж. М .; Али, А .; Хиллер, М.С. Варенье. Chem. Soc., 1999, 121, 866-867. (Дои:10.1021 / ja9829259 )
- ^ Kende, A. S .; Kawamura, K .; ДеВита, Р. Дж. Варенье. Chem. Soc., 1990, 112 4070-4072. (Дои:10.1021 / ja00166a072 ).
- ^ Kende, A. S., Koch, K .; Дори, G .; Kaldor, I .; Лю, К. Варенье. Chem. Soc., 1993, 115, 9842-9843. (Дои:10.1021 / ja00074a078 ).
- ^ Хонг, К. И, Киши, Ю. Варенье. Chem. Soc., 1991, 113, 9693-9694. (Дои:10.1021 / ja00025a056 ).
- ^ Tanimoto, N .; Герриц, С. У .; Sawabe, A .; Нода, Т .; Filla, S.A .; Масамунэ, С. Энгью. Chem. Int. Эд., 2003, 33, 673-675. (Дои:10.1002 / anie.199406731 ).
- ^ Эванс, Д. А .; Блэк, У.С. Варенье. Chem. Soc., 1993, 115, 4497-4513. (Дои:10.1021 / ja00064a011 ).
- ^ Tang, W .; Прусов, Э.В. Орг. Lett., 2012, 14 4690-4693. (Дои:10.1021 / ol302219x ).
- ^ Coleman, R. S .; Walczak, M.C .; Кэмпбелл, Э. Варенье. Chem. Soc., 2005, 127, 16036-16039. (Дои:10.1021 / ja056217g ).
- ^ Roth, G.P .; Фарина, В .; Либескинд, Л. С .; Пенья-Кабрера, Э. Tetrahedron Lett. 1995, 36, 2191.
- ^ Renaldo, A. F .; Labadie, J. W .; Стилле, Дж. К. Органический синтез, Сб. Vol. 8, стр. 268 (1993); Vol. 67, стр.86 (1989). (Статья )
- ^ Стилле-реакции с тетраалкилстаннанами и фенилтриалкилстаннанами в легкоплавких смесях сахар-мочевина-сольДжованни Императо, Рудольф Васольд, Буркхард Кениг Расширенный синтез и катализ Том 348, выпуск 15, страницы 2243–47 2006 Дои:10.1002 / adsc.2006
- ^ П. Эспине, А. М. Эчаваррен (2004). «Механизмы реакции стилля». Angewandte Chemie International Edition. 43 (36): 4704–4734. Дои:10.1002 / anie.200300638. PMID 15366073.
- ^ Knight, S.D .; Overman, L.E .; Пайродо, Г. Варенье. Chem. Soc., 1993, 115, 9293-9294. (Дои:10.1021 / ja00073a057 )
- ^ Monera, E .; Ортар, Г. Биорг. Med. Chem. Lett., 2000, 10, 1815-1818. (Дои:10.1016 / S0960-894X (00) 00344-9 ).
- ^ Gyorkos, A.C .; Stille, J. K .; Хегедус, Л.С. Варенье. Chem. Soc., 1990, 112, 8465-8472. (Дои:10.1021 / ja00179a035 ).
- ^ Yue, W. S .; Ли, Дж. Дж. Орг. Lett., 2002, 4, 2201-2203. (Дои:10.1021 / ol0260425 )
внешняя ссылка
- Раздаточный материал реакции Стилла из группы Майерс.
- Стилле реакция на organic-chemistry.org
- Реакция Стилле - Синтетические протоколы из organic-reaction.com