Алгоритм Смита – Уотермана - Smith–Waterman algorithm
| Класс | Выравнивание последовательности |
|---|---|
| Худший случай спектакль | |
| Худший случай космическая сложность |


В Алгоритм Смита – Уотермана выполняет местные выравнивание последовательностей; то есть для определения похожих областей между двумя строками последовательности нуклеиновых кислот или белковые последовательности. Вместо того, чтобы смотреть на весь последовательность, алгоритм Смита – Уотермана сравнивает сегменты всех возможных длин и оптимизирует то мера сходства.
Алгоритм был впервые предложен Темпл Ф. Смит и Майкл С. Уотерман в 1981 г.[1] Словно Алгоритм Нидлмана – Вунша, из которых это вариация, Смит – Уотерман является динамическое программирование алгоритм. Таким образом, он обладает желаемым свойством, заключающимся в том, что он гарантирует оптимальное локальное выравнивание по отношению к используемой системе подсчета очков (которая включает матрица замещения и скоринг схема). Основное отличие от Алгоритм Нидлмана – Вунша заключается в том, что ячейки матрицы отрицательной оценки устанавливаются в ноль, что делает видимыми локальные выравнивания (таким образом, положительные оценки). Процедура отслеживания начинается с ячейки матрицы с наивысшей оценкой и продолжается до тех пор, пока не встретится ячейка с нулевой оценкой, что дает локальное выравнивание с самой высокой оценкой. Из-за своей квадратичной сложности во времени и пространстве он часто не может быть практически применен к крупномасштабным задачам и заменяется менее общими, но более эффективными в вычислительном отношении альтернативами, такими как (Gotoh, 1982),[2] (Альтшул и Эриксон, 1986),[3] и (Майерс и Миллер, 1988).[4]
История
В 1970 году Сол Б. Нидлман и Кристиан Д. Вунш предложили эвристический алгоритм гомологии для выравнивания последовательностей, также известный как алгоритм Нидлмана – Вунша.[5] Это алгоритм глобального выравнивания, который требует шаги расчета ( и - длины двух выравниваемых последовательностей). Он использует итеративное вычисление матрицы для демонстрации глобального выравнивания. В следующее десятилетие Санкофф,[6] Райхерт,[7] Бейер[8] и другие сформулировали альтернативные эвристические алгоритмы для анализа последовательностей генов. Продавцы представили систему измерения расстояний последовательности.[9] В 1976 году Waterman et al. добавили концепцию зазоров в исходную систему измерения.[10] В 1981 году Смит и Уотерман опубликовали свой алгоритм Смита – Уотермана для расчета локального выравнивания.


Алгоритм Смита – Уотермана довольно требователен ко времени: чтобы выровнять две последовательности длины и , требуется время. Gotoh[2] и Альтшул[3] оптимизировал алгоритм, чтобы шаги. Сложность пространства была оптимизирована Майерсом и Миллером.[4] от к (линейный), где - длина более короткой последовательности для случая, когда требуется только одно из многих возможных оптимальных выравниваний.

Мотивация
За последние годы, геномные проекты На множестве организмов было получено огромное количество данных о последовательностях генов и белков, что требует вычислительного анализа. Выравнивание последовательностей показывает отношения между генами или между белками, что позволяет лучше понять их гомологию и функциональность. Выравнивание последовательностей также может выявить сохраненные домены и мотивы.
Одним из мотивов для локального выравнивания является сложность получения правильных выравниваний в областях с низким сходством между отдаленно связанными биологическими последовательностями, потому что мутации добавили слишком много «шума» за время эволюции, чтобы можно было провести значимое сравнение этих областей. Локальное выравнивание полностью избегает таких областей и фокусируется на тех, которые имеют положительный результат, то есть на тех, которые имеют эволюционно сохраненный сигнал сходства. Предпосылкой для локального согласования является отрицательная оценка ожидания. Оценка ожидания определяется как средняя оценка, которую система оценок (матрица замещения и штрафы за разрыв ) даст случайную последовательность.
Еще одна мотивация для использования локальных выравниваний заключается в том, что существует надежная статистическая модель (разработанная Карлином и Альтшулом) для оптимальных локальных выравниваний. Выравнивание неродственных последовательностей имеет тенденцию давать оптимальные баллы локального выравнивания, которые следуют распределению экстремальных значений. Это свойство позволяет программам создавать ожидаемое значение для оптимального локального выравнивания двух последовательностей, которое является мерой того, как часто две несвязанные последовательности будут давать оптимальное локальное выравнивание, оценка которого больше или равна наблюдаемой оценке. Очень низкие ожидаемые значения указывают на то, что две рассматриваемые последовательности могут быть гомологичный, что означает, что у них может быть общий предок.
Алгоритм
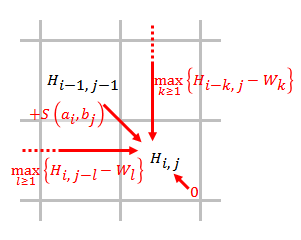
Позволять и быть выравниваемыми последовательностями, где и длины и соответственно.




- Определите матрицу замены и схему штрафа за пропуск.
- - Оценка сходства элементов, составляющих две последовательности
- - Штраф за разрыв, имеющий длину
- Постройте матрицу оценок и инициализируйте его первую строку и первый столбец. Размер оценочной матрицы . В матрице используется индексирование на основе 0.
- Заполните оценочную матрицу, используя приведенное ниже уравнение.
- где
- это оценка согласования и ,
- это оценка, если находится в конце промежутка длины ,
- это оценка, если находится в конце промежутка длины ,
- означает, что сходства нет до и .
- Проследить. Начиная с наивысшего результата в матрице оценок и заканчивая ячейкой матрицы, которая имеет оценку 0, обратная трассировка на основе источника каждой оценки рекурсивно для создания наилучшего локального выравнивания.














Объяснение
Алгоритм Смита – Уотермана выравнивает две последовательности по совпадениям / несовпадениям (также известным как замены), вставкам и удалениям. И вставки, и удаления - это операции, при которых появляются пробелы, которые обозначаются тире. Алгоритм Смита – Уотермана состоит из нескольких шагов:
- Определите матрицу замены и схему штрафа за пропуск. Матрица замещения присваивает каждой паре оснований или аминокислот оценку совпадения или несоответствия. Обычно совпадения получают положительные оценки, а несоответствия - относительно низкие. Функция штрафа за пропуск определяет стоимость очков за открытие или расширение пропусков. Предлагается, чтобы пользователи выбирали подходящую систему подсчета очков в зависимости от целей. Кроме того, рекомендуется пробовать различные комбинации матриц замещения и штрафов за пробелы.
- Инициализировать скоринговую матрицу. Размеры оценочной матрицы равны 1 + длина каждой последовательности соответственно. Все элементы первой строки и первого столбца устанавливаются в 0. Дополнительная первая строка и первый столбец позволяют выровнять одну последовательность по другой в любой позиции, а установка их в 0 освобождает конечный пробел от штрафа.
- Подсчет очков. Оценивайте каждый элемент слева направо, сверху вниз в матрице, учитывая результаты замен (диагональные баллы) или добавления пробелов (горизонтальные и вертикальные баллы). Если ни одна из оценок не является положительной, этому элементу присваивается 0. В противном случае используется наивысшая оценка и записывается источник этой оценки.
- Проследить. Начиная с элемента с наивысшей оценкой, выполняется обратная трассировка на основе источника каждой оценки рекурсивно, пока не будет обнаружен 0. В этом процессе генерируются сегменты, которые имеют наивысшую оценку сходства на основе данной системы оценок. Чтобы получить второе лучшее локальное выравнивание, примените процесс трассировки, начиная со второго наивысшего результата за пределами следа наилучшего выравнивания.
Сравнение с алгоритмом Нидлмана – Вунша.
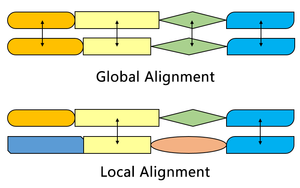
Алгоритм Смита – Уотермана находит сегменты в двух последовательностях, которые имеют сходство, в то время как алгоритм Нидлмана – Вунша выравнивает две полные последовательности. Следовательно, они служат разным целям. Оба алгоритма используют концепции матрицы подстановки, функции штрафа за пробелы, матрицы оценки и процесса отслеживания. Три основных отличия:
| Алгоритм Смита – Уотермана | Алгоритм Нидлмана – Вунша | |
|---|---|---|
| Инициализация | Первая строка и первый столбец установлены на 0 | Первая строка и первый столбец подлежат штрафу за пробел |
| Подсчет очков | Отрицательная оценка установлена на 0 | Оценка может быть отрицательной |
| Проследить | Начните с наивысшего результата, закончите, когда встретится 0 | Начните с ячейки в правом нижнем углу матрицы, закончите в верхней левой ячейке |
Одно из наиболее важных отличий заключается в том, что в системе оценок алгоритма Смита – Уотермана не присваивается отрицательная оценка, что обеспечивает локальное выравнивание. Когда какой-либо элемент имеет оценку ниже нуля, это означает, что последовательности до этой позиции не имеют сходства; затем этот элемент будет установлен в ноль, чтобы исключить влияние предыдущего выравнивания. Таким образом, вычисление может продолжить поиск выравнивания в любой позиции впоследствии.
Исходная оценочная матрица алгоритма Смита – Уотермана позволяет выровнять любой сегмент одной последовательности с произвольным положением в другой последовательности. Однако в алгоритме Нидлмана – Вунша необходимо учитывать штраф за разрыв в конце, чтобы выровнять полные последовательности.
Матрица замещения
Каждой замене основания или аминокислотной замене присваивается балл. Как правило, совпадениям присваиваются положительные оценки, а несоответствиям - относительно более низкие оценки. Возьмем для примера последовательность ДНК. Если совпадения получают +1, несоответствия получают -1, тогда матрица подстановки:
| А | г | C | Т | |
|---|---|---|---|---|
| А | 1 | -1 | -1 | -1 |
| г | -1 | 1 | -1 | -1 |
| C | -1 | -1 | 1 | -1 |
| Т | -1 | -1 | -1 | 1 |
Эта матрица замещения может быть описана как:

Различные замены оснований или аминокислотные замены могут иметь разные оценки. Матрица замещения аминокислот обычно сложнее, чем у оснований. Увидеть PAM, BLOSUM.
Штраф за разрыв
Штраф за пробел обозначает баллы за вставку или удаление. Простая стратегия штрафа за пробелы заключается в использовании фиксированной оценки для каждого пробела. В биологии, однако, по практическим соображениям необходимо подсчитывать баллы иначе. С одной стороны, частичное сходство между двумя последовательностями - обычное явление; с другой стороны, событие мутации одного гена может привести к вставке одного длинного промежутка. Поэтому соединенные промежутки, образующие длинный промежуток, обычно более предпочтительны, чем множественные рассеянные короткие промежутки. Чтобы учесть это различие, в систему подсчета очков были добавлены концепции открытия и увеличения промежутка. Оценка открытия разрыва обычно выше, чем оценка расширения разрыва. Например, параметр по умолчанию в EMBOSS Вода составляют: открытие зазора = 10, расширение зазора = 0,5.
Здесь мы обсуждаем две общие стратегии штрафа за пробелы. Увидеть Штраф за разрыв для большего количества стратегий. - функция штрафа за пробел для пробела длиной :
Линейный
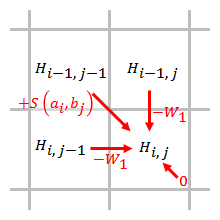
Штраф за линейный разрыв имеет одинаковые баллы за открытие и расширение разрыва:
,

где стоимость разового пробела.

Штраф за разрыв прямо пропорционален длине зазора. При использовании линейного штрафа за разрыв алгоритм Смита – Уотермана можно упростить до:

Упрощенный алгоритм использует шаги. Когда элемент оценивается, необходимо учитывать только штрафы за пробелы от элементов, которые непосредственно примыкают к этому элементу.
Аффинный
Штраф за аффинный разрыв учитывает открытие и расширение разрыва отдельно:
,

где - штраф за открытие промежутка, и штраф за удлинение промежутка. Например, штраф за разрыв длиной 2 составляет .



В исходной статье алгоритма Смита – Уотермана использовался произвольный штраф за пропуски. Оно использует шагов, поэтому требует времени. Гото оптимизировал шаги для штрафа за аффинный пробел, чтобы ,[2] но оптимизированный алгоритм пытается найти только одно оптимальное выравнивание, и не гарантируется, что оптимальное выравнивание будет найдено.[3] Альтшул модифицировал алгоритм Гото, чтобы найти все оптимальные выравнивания, сохранив при этом вычислительную сложность.[3] Позже Майерс и Миллер указали, что алгоритм Гото и Альтшула может быть дополнительно модифицирован на основе метода, опубликованного Хиршбергом в 1975 г.[11] и применил этот метод.[4] Алгоритм Майерса и Миллера может выровнять две последовательности, используя пространство, с длина более короткой последовательности.

Пример штрафа за разрыв
Возьмите выравнивание последовательностей TACGGGCCCGCTAC и TAGCCCTATCGGTCA Например, при использовании функции линейного штрафа за разрыв результат будет (Выравнивания выполняются с помощью EMBOSS Water. Матрица замещения - DNAful. Размер раскрытия и расширения зазора равен 1,0):
TACGGGCCCGCTA-C|| | || ||| |TA --- G-CC-CTATC
Когда используется штраф за аффинный пробел, результат будет (открытие и расширение пропуска - 5,0 и 1,0 соответственно):
TACGGGCCCGCTA|| ||| |||TA --- GCC - CTA
Этот пример показывает, что штраф за аффинный пробел может помочь избежать разбросанных небольших пробелов.
Матрица оценок
Функция скоринговой матрицы заключается в проведении однозначных сравнений между всеми компонентами в двух последовательностях и записи оптимальных результатов выравнивания. Процесс выставления оценок отражает концепцию динамического программирования. Окончательное оптимальное выравнивание находится путем итеративного расширения растущего оптимального выравнивания. Другими словами, текущее оптимальное выравнивание генерируется путем решения, какой путь (совпадение / несоответствие или вставка пробела) дает наивысший балл из предыдущего оптимального выравнивания. Размер матрицы равен длине одной последовательности плюс 1 на длину другой последовательности плюс 1. Дополнительная первая строка и первый столбец служат для выравнивания одной последовательности с любыми позициями в другой последовательности. И первая строка, и первый столбец установлены на 0, так что зазор в конце не штрафуется. Матрица начальных оценок:
| б1 | … | бj | … | бм | ||
|---|---|---|---|---|---|---|
| 0 | 0 | … | 0 | … | 0 | |
| а1 | 0 | |||||
| … | … | |||||
| ая | 0 | |||||
| … | … | |||||
| ап | 0 |
пример
Сделайте выравнивание последовательностей ДНК TGTTACGG и GGTTGACTA Например. Используйте следующую схему:
- Матрица замещения:
- Штраф за разрыв: (штраф за линейный разрыв )



Инициализируйте и заполните матрицу оценок, как показано ниже. На этом рисунке показан процесс оценки первых трех элементов. Желтым цветом обозначены рассматриваемые базы. Красный цвет указывает на максимально возможную оценку для оцениваемой ячейки.

Готовая оценочная матрица показана ниже слева. Синий цвет показывает наивысший балл. Элемент может получить оценку от более чем одного элемента, каждый из них будет формировать свой путь, если этот элемент прослеживается. В случае нескольких наивысших оценок, обратное отслеживание должно выполняться, начиная с каждой наивысшей оценки. Процесс отслеживания показан ниже справа. Лучшее локальное выравнивание создается в обратном направлении.
 | 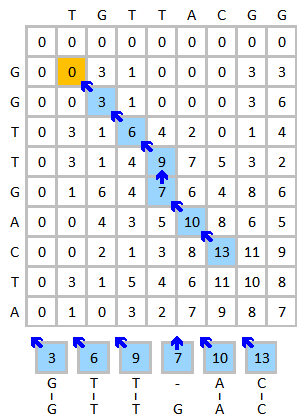 |
| Матрица завершена (наивысший балл отмечен синим цветом) | Процесс отслеживания и результат согласования |
Результат совмещения:
Г Т Т - А С| | | | |G T T G A C
Реализация
Реализация алгоритма Смита – Уотермана SSEARCH доступна в ФАСТА пакет анализа последовательности от UVA FASTA Загрузки. Эта реализация включает Altivec ускоренный код для PowerPC Процессоры G4 и G5, которые ускоряют сравнение в 10–20 раз, используя модификацию подхода Wozniak, 1997,[12] и векторизация SSE2, разработанная Фарраром[13] создание оптимального белка база данных последовательностей поиски вполне практичные. Библиотека SSW расширяет реализацию Фаррара, возвращая информацию о выравнивании в дополнение к оптимальной оценке Смита – Уотермана.[14]
Ускоренные версии
FPGA
Cray продемонстрировали ускорение алгоритма Смита – Уотермана с использованием реконфигурируемые вычисления платформа на основе FPGA микросхем, с результатами, показывающими увеличение скорости до 28 раз по сравнению со стандартными решениями на базе микропроцессоров. Другая версия алгоритма Смита – Уотермана на основе FPGA показывает ускорение FPGA (Virtex-4) до 100 раз.[15] над процессором Opteron с тактовой частотой 2,2 ГГц.[16] В TimeLogic Системы DeCypher и CodeQuest также ускоряют поиск Smith – Waterman и Framesearch с помощью карт PCIe FPGA.
Магистерская работа 2011 г. [17] включает анализ ускорения Смита – Уотермана на основе ПЛИС.
В публикации 2016 г. Код OpenCL, скомпилированный с помощью Xilinx SDAccel, ускоряет секвенирование генома, в 12-21 раз превосходит производительность CPU / GPU / Вт, была представлена очень эффективная реализация. При использовании одной карты PCIe FPGA, оснащенной FPGA Xilinx Virtex-7 2000T, производительность на ваттный уровень была выше, чем у CPU / GPU в 12-21 раз.
GPU
Национальная лаборатория Лоуренса Ливермора и Министерства энергетики США (США) Объединенный институт генома реализовала ускоренную версию поиска локального выравнивания последовательностей Смита – Уотермана с использованием графические процессоры (GPU) с предварительными результатами, показывающими двукратное ускорение по сравнению с программными реализациями.[18] Подобный метод уже реализован в программе Biofacet с 1997 года с тем же коэффициентом ускорения.[19]
Несколько GPU реализации алгоритма в NVIDIA с CUDA Платформа C.[20] По сравнению с наиболее известной реализацией ЦП (с использованием инструкций SIMD на архитектуре x86) Фарраром, тесты производительности этого решения с использованием одного NVidia GeForce 8800 GTX card показывают небольшое увеличение производительности для небольших последовательностей, но небольшое снижение производительности для более крупных. Однако те же тесты, запущенные на двойном NVidia GeForce 8800 GTX карты почти в два раза быстрее, чем реализация Farrar для всех протестированных размеров последовательностей.
Теперь доступна новая реализация SW CUDA на графическом процессоре, которая работает быстрее, чем предыдущие версии, а также снимает ограничения на длину запросов. Увидеть CUDASW ++.
Сообщалось об одиннадцати различных реализациях ПО на CUDA, три из которых сообщают об ускорении в 30 раз.[21]
SIMD
В 2000 году была произведена быстрая реализация алгоритма Смита – Уотермана с использованием SIMD технология доступна в Intel Pentium MMX Процессоры и аналогичные технологии были описаны в публикации Rognes и Seeberg.[22] В отличие от подхода Возняка (1997), новая реализация была основана на векторах, параллельных последовательности запроса, а не на диагональных векторах. Компания Sencel Bioinformatics подал заявку на патент, охватывающий этот подход. Sencel продолжает разработку программного обеспечения и бесплатно предоставляет исполняемые файлы для академического использования.
А SSE2 В настоящее время доступна векторизация алгоритма (Farrar, 2007), обеспечивающая ускорение в 8-16 раз на процессорах Intel / AMD с расширениями SSE2.[13] При работе на процессоре Intel с использованием Основная микроархитектура реализация SSE2 достигает 20-кратного увеличения. Реализация SSE2 Фаррара доступна в виде программы SSEARCH в ФАСТА Пакет сравнения последовательностей. SSEARCH включен в Европейский институт биоинформатики набор программы поиска сходства.
Датская биоинформатическая компания CLC биография достигла ускорения почти на 200 по сравнению со стандартной программной реализацией с SSE2 на процессоре Intel Core 2 Duo 2,17 ГГц, согласно данным общедоступный технический документ.
Ускоренная версия алгоритма Смита – Уотермана на Intel и AMD на базе серверов Linux, поддерживается GenCore 6 пакет, предлагаемый Биоскелерация. Тесты производительности этого программного пакета показывают увеличение скорости до 10 раз по сравнению со стандартной программной реализацией на том же процессоре.
В настоящее время единственная компания в области биоинформатики, предлагающая решения как SSE, так и FPGA, ускоряющие Smith – Waterman, CLC биография добился ускорения более чем в 110 раз по сравнению со стандартными реализациями программного обеспечения с Куб биоинформатики CLC[нужна цитата ]
Самая быстрая реализация алгоритма на процессорах с SSSE3 можно найти программное обеспечение SWIPE (Rognes, 2011),[23] который доступен под Стандартная общественная лицензия GNU Affero. Параллельно это программное обеспечение сравнивает остатки из шестнадцати различных последовательностей базы данных с одним остатком запроса. Используя последовательность запросов остатков 375, скорость обновления 106 миллиардов ячеек в секунду (GCUPS) была достигнута на двойном Intel Xeon Шестиядерная процессорная система X5650, которая более чем в шесть раз быстрее, чем программное обеспечение, основанное на «полосатом» подходе Фаррара. Это быстрее чем ВЗРЫВ при использовании матрицы BLOSUM50.
Также существует diagonalsw, реализация алгоритма Смита – Уотермана на языках C и C ++ с наборами инструкций SIMD (SSE4.1 для платформы x86 и AltiVec для платформы PowerPC). Он находится под лицензией MIT с открытым исходным кодом.
Ячейка широкополосного доступа
В 2008 году Фаррар[24] описал порт Полосатого Смита – Уотермана[13] к Ячейка широкополосного доступа и сообщил о скорости 32 и 12 GCUPS на Блейд-сервер IBM QS20 и Sony PlayStation 3 соответственно.
Ограничения
Быстрое распространение генетических данных бросает вызов скорости текущих алгоритмов выравнивания последовательностей ДНК. Существенные потребности в эффективном и точном методе обнаружения вариантов ДНК требуют инновационных подходов для параллельной обработки в реальном времени. Оптические вычисления подходы были предложены в качестве многообещающей альтернативы текущим электрическим реализациям. OptCAM является примером таких подходов и показывает, что он быстрее, чем алгоритм Смита – Уотермана.[25]
Смотрите также
- Биоинформатика
- Выравнивание последовательности
- Последовательный майнинг
- Алгоритм Нидлмана – Вунша
- Расстояние Левенштейна
- ВЗРЫВ
- ФАСТА
использованная литература
- ^ Смит, Темпл Ф. и Уотерман, Майкл С. (1981). «Идентификация общих молекулярных подпоследовательностей» (PDF). Журнал молекулярной биологии. 147 (1): 195–197. CiteSeerX 10.1.1.63.2897. Дои:10.1016/0022-2836(81)90087-5. PMID 7265238.
- ^ а б c Осаму Гото (1982). «Улучшенный алгоритм сопоставления биологических последовательностей». Журнал молекулярной биологии. 162 (3): 705–708. CiteSeerX 10.1.1.204.203. Дои:10.1016/0022-2836(82)90398-9. PMID 7166760.
- ^ а б c d Стивен Ф. Альтшул и Брюс В. Эриксон (1986). «Оптимальное выравнивание последовательностей с использованием затрат аффинного разрыва». Вестник математической биологии. 48 (5–6): 603–616. Дои:10.1007 / BF02462326. PMID 3580642. S2CID 189889143.
- ^ а б c Миллер, Уэбб; Майерс, Юджин (1988). «Оптимальные соосности в линейном пространстве». Биоинформатика. 4 (1): 11–17. CiteSeerX 10.1.1.107.6989. Дои:10.1093 / биоинформатика / 4.1.11. PMID 3382986.
- ^ Саул Б. Нидлман; Кристиан Д. Вунш (1970). «Общий метод, применимый к поиску сходства в аминокислотной последовательности двух белков». Журнал молекулярной биологии. 48 (3): 443–453. Дои:10.1016/0022-2836(70)90057-4. PMID 5420325.
- ^ Санкофф Д. (1972). «Соответствующие последовательности при ограничениях на удаление / вставку». Труды Национальной академии наук Соединенных Штатов Америки. 69 (1): 4–6. Bibcode:1972ПНАС ... 69 .... 4С. Дои:10.1073 / pnas.69.1.4. ЧВК 427531. PMID 4500555.
- ^ Томас А. Райхерт; Дональд Н. Коэн; Эндрю К. Вонг (1973). «Применение теории информации к генетическим мутациям и сопоставлению полипептидных последовательностей». Журнал теоретической биологии. 42 (2): 245–261. Дои:10.1016 / 0022-5193 (73) 90088-X. PMID 4762954.
- ^ Уильям А. Бейер, Майрон Л. Стейн, Темпл Ф. Смит и Станислав М. Улам (1974). «Метрика молекулярной последовательности и деревья эволюции». Математические биологические науки. 19 (1–2): 9–25. Дои:10.1016/0025-5564(74)90028-5.CS1 maint: несколько имен: список авторов (ссылка на сайт)
- ^ Питер Х. Селлерс (1974). «К теории и вычислению эволюционных расстояний». Журнал прикладной математики. 26 (4): 787–793. Дои:10.1137/0126070.
- ^ М.С. Уотерман; T.F Smith; В. А. Бейер (1976). «Некоторые показатели биологической последовательности». Успехи в математике. 20 (3): 367–387. Дои:10.1016/0001-8708(76)90202-4.
- ^ Д. С. Хиршберг (1975). «Алгоритм линейного пространства для вычисления максимальных общих подпоследовательностей». Коммуникации ACM. 18 (6): 341–343. CiteSeerX 10.1.1.348.4774. Дои:10.1145/360825.360861. S2CID 207694727.
- ^ Возняк, Анджей (1997). «Использование видео-ориентированных инструкций для ускорения сравнения последовательностей» (PDF). Компьютерные приложения в биологических науках (CABIOS). 13 (2): 145–50. Дои:10.1093 / биоинформатика / 13.2.145. PMID 9146961.
- ^ а б c Фаррар, Майкл С. (2007). «Полосатый Смит – Уотерман ускоряет поиск в базе данных в шесть раз по сравнению с другими реализациями SIMD» (PDF). Биоинформатика. 23 (2): 156–161. Дои:10.1093 / биоинформатика / btl582. PMID 17110365.
- ^ Чжао, Мэнъяо; Ли, Ван-Пин; Гаррисон, Эрик П. Март, Габор Т. (4 декабря 2013 г.). «Библиотека SSW: библиотека SIMD Smith-Waterman C / C ++ для использования в геномных приложениях». PLOS ONE. 8 (12): e82138. arXiv:1208.6350. Bibcode:2013PLoSO ... 882138Z. Дои:10.1371 / journal.pone.0082138. ЧВК 3852983. PMID 24324759.
- ^ Статьи о FPGA 100x: «Архивная копия» (PDF). Архивировано из оригинал (PDF) на 2008-07-05. Получено 2007-10-17.CS1 maint: заархивированная копия как заголовок (ссылка на сайт), «Архивная копия» (PDF). Архивировано из оригинал (PDF) на 2008-07-05. Получено 2007-10-17.CS1 maint: заархивированная копия как заголовок (ссылка на сайт), и «Архивная копия» (PDF). Архивировано из оригинал (PDF) на 2011-07-20. Получено 2007-10-17.CS1 maint: заархивированная копия как заголовок (ссылка на сайт)
- ^ Progeniq Pte. ООО "Информационный документ - Ускорение интенсивных приложений в 10–50 раз для устранения узких мест в вычислительных рабочих процессах ".
- ^ Вермий, Эрик (2011). Выравнивание генетических последовательностей на суперкомпьютерной платформе (PDF) (Кандидатская диссертация). Делфтский технологический университет. Архивировано из оригинал (PDF) на 2011-09-30. Получено 2011-08-17.
- ^ Лю, Ян; Хуанг, Уэйн; Джонсон, Джон; Вайдья, Шейла (2006). Смит – Уотерман с ускорением на GPU. Конспект лекций по информатике. 3994. SpringerLink. стр.188–195. Дои:10.1007/11758549_29. ISBN 978-3-540-34385-1.
- ^ «Биоинформатика, поиск и анализ последовательности с высокой пропускной способностью (технический документ)». GenomeQuest. Архивировано из оригинал 13 мая 2008 г.. Получено 2008-05-09.
- ^ «Зона CUDA». Nvidia. Получено 2010-02-25.
- ^ Рогнес, Торбьёрн и Сиберг, Эрлинг (2000). «Шестикратное ускорение поиска в базе данных последовательностей Смита – Уотермана с использованием параллельной обработки на обычных микропроцессорах» (PDF). Биоинформатика. 16 (8): 699–706. Дои:10.1093 / биоинформатика / 16.8.699. PMID 11099256.
- ^ Рогнес, Торбьёрн (2011). «Ускоренный поиск в базе данных Смита – Уотермана с распараллеливанием между последовательностями SIMD». BMC Bioinformatics. 12: 221. Дои:10.1186/1471-2105-12-221. ЧВК 3120707. PMID 21631914.
- ^ Фаррар, Майкл С. (2008). «Оптимизация Смита – Ватермана для механизма сотовой широкополосной связи». Архивировано из оригинал на 2012-02-12. Цитировать журнал требует
| журнал =(Помогите) - ^ Малеки, Эхсан; Кухи, Сомайе; Кавехваш, Захра; Машаги, Алиреза (2020). «OptCAM: сверхбыстрая полностью оптическая архитектура для обнаружения вариантов ДНК». Журнал биофотоники. 13 (1): e201900227. Дои:10.1002 / jbio.201900227. PMID 31397961.
внешние ссылки
- JAligner - реализация алгоритма Смита – Уотермана на Java с открытым исходным кодом
- B.A.B.A. - апплет (с исходным кодом), который наглядно объясняет алгоритм
- FASTA / SSEARCH - страница услуг на сайте EBI
- Плагин UGENE Smith – Waterman - реализация алгоритма с открытым исходным кодом, совместимая с SSEARCH, с графическим интерфейсом, написанным на C ++
- ОПАЛ - библиотека SIMD C / C ++ для массового оптимального выравнивания последовательностей
- диагональw - реализация C / C ++ с открытым исходным кодом с наборами инструкций SIMD (особенно SSE4.1) под лицензией MIT
- SSW - библиотека C ++ с открытым исходным кодом, предоставляющая API для реализации SIMD алгоритма Смита – Уотермана по лицензии MIT
- выравнивание мелодической последовательности - реализация javascript для выравнивания мелодических последовательностей