Последовательность нуклеиновой кислоты - Nucleic acid sequence
Эта статья нужны дополнительные цитаты для проверка. (Март 2014 г.) (Узнайте, как и когда удалить этот шаблон сообщения) |
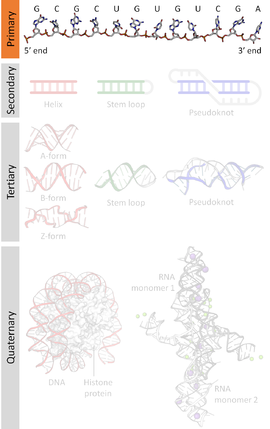
А последовательность нуклеиновой кислоты представляет собой последовательность оснований, обозначенную серией из пяти разных букв, которые указывают порядок нуклеотиды формирование аллели в пределах ДНК (используя GACT) или РНК (GACU) молекула. По соглашению последовательности обычно представляются из 5 футов до конца 3. Для ДНК смысл прядь используется. Поскольку нуклеиновые кислоты обычно линейны (неразветвленные) полимеры, указание последовательности эквивалентно определению ковалентный структура всей молекулы. По этой причине последовательность нуклеиновой кислоты также называется первичная структура.
Последовательность может представлять Информация. Биологическая дезоксирибонуклеиновая кислота представляет собой информацию, которая управляет функциями живого существа.
Нуклеиновые кислоты также имеют вторичная структура и третичная структура. Первичную структуру иногда ошибочно называют первичная последовательность. И наоборот, не существует параллельной концепции вторичной или третичной последовательности.
Нуклеотиды
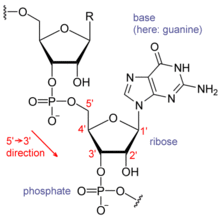

Нуклеиновые кислоты состоят из цепочки связанных звеньев, называемых нуклеотидами. Каждый нуклеотид состоит из трех субъединиц: фосфат группа и сахар (рибоза в случае РНК, дезоксирибоза в ДНК ) составляют основу нити нуклеиновой кислоты, а прикрепленный к сахару один из множества азотистые основания. Азотистые основания важны в базовая пара прядей для формирования более высокого уровня вторичный и третичная структура такие как знаменитый двойная спираль.
Возможные буквы А, C, грамм, и Т, представляющие четыре нуклеотид базы нити ДНК - аденин, цитозин, гуанин, тимин – ковалентно связан с фосфодиэфир позвоночник. В типичном случае последовательности печатаются вплотную друг к другу без пропусков, как в последовательности AAAGTCTGAC, читаемой слева направо в От 5 футов до 3 футов направление. Что касается транскрипция, последовательность находится на кодирующей цепи, если она имеет тот же порядок, что и транскрибируемая РНК.
Одна последовательность может быть дополнительный в другую последовательность, что означает, что они имеют основу в каждой позиции в дополнительной (то есть от A до T, от C до G) и в обратном порядке. Например, дополнительная последовательность к TTAC - GTAA. Если одна цепь двухцепочечной ДНК считается смысловой цепью, тогда другая цепь, считающаяся антисмысловой цепью, будет иметь последовательность, комплементарную смысловой цепи.
Обозначение
Сравнение и определение% разницы между двумя нуклеотидными последовательностями.
- AAТCCGCТЕГ
- AAАCCCTТЕГ
- Учитывая две 10-нуклеотидные последовательности, выровняйте их и сравните различия между ними. Вычислите процент сходства, разделив количество различных оснований ДНК на общее количество нуклеотидов. В приведенном выше случае есть три различия в последовательности из 10 нуклеотидов. Следовательно, разделите 7/10, чтобы получить 70% сходства, и вычтите это из 100%, чтобы получить разницу в 30%.
Хотя A, T, C и G обозначают конкретный нуклеотид в позиции, есть также буквы, которые обозначают неоднозначность, которые используются, когда в этом положении может встречаться более одного вида нуклеотидов. Правила Международного союза теоретической и прикладной химии (ИЮПАК ) являются следующими:[1]
| Символ[2] | Описание | Представленные базы | Дополнение | ||||
|---|---|---|---|---|---|---|---|
| А | АDenine | А | 1 | Т | |||
| C | Cиттозин | C | грамм | ||||
| грамм | граммуанин | грамм | C | ||||
| Т | Тгимин | Т | А | ||||
| U | Uрасил | U | А | ||||
| W | WEak | А | Т | 2 | W | ||
| S | Sсильный | C | грамм | S | |||
| M | аMя не | А | C | K | |||
| K | Kэто | грамм | Т | M | |||
| р | пурине | А | грамм | Y | |||
| Y | пYримидин | C | Т | р | |||
| B | не А (B идет после A) | C | грамм | Т | 3 | V | |
| D | не C (D идет после C) | А | грамм | Т | ЧАС | ||
| ЧАС | не G (ЧАС идет после G) | А | C | Т | D | ||
| V | не Т (V идет после T и U) | А | C | грамм | B | ||
| N | любой Nуклеотид (не пробел) | А | C | грамм | Т | 4 | N |
| Z | Zэро | 0 | Z | ||||
Эти символы также действительны для РНК, за исключением U (урацил), заменяющего T (тимин).[1]
Помимо аденина (A), цитозина (C), гуанина (G), тимина (T) и урацила (U), ДНК и РНК также содержат основания, которые были модифицированы после образования цепи нуклеиновой кислоты. В ДНК наиболее распространенным модифицированным основанием является 5-метилцитидин (m5C). В РНК есть много модифицированных оснований, включая псевдоуридин (Ψ), дигидроуридин (D), инозин (I), риботимидин (rT) и 7-метилгуанозин (m7G).[3][4] Гипоксантин и ксантин две из многих баз, созданных с помощью мутаген наличие, их обоих посредством дезаминирования (замены аминогруппы карбонильной группой). Гипоксантин производится из аденин, а ксантин производится из гуанин.[5] Аналогичным образом дезаминирование цитозин приводит к урацил.
Биологическое значение
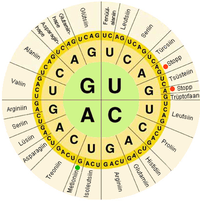
В биологических системах нуклеиновые кислоты содержат информацию, которая используется живым организмом. клетка построить конкретные белки. Последовательность азотистые основания на цепи нуклеиновой кислоты переведено клеточными механизмами в последовательность аминокислоты составление белковой нити. Каждая группа из трех баз, называемая кодон, соответствует одной аминокислоте, и существует конкретная генетический код при котором каждая возможная комбинация из трех оснований соответствует определенной аминокислоте.
В центральная догма молекулярной биологии описывает механизм построения белков с использованием информации, содержащейся в нуклеиновых кислотах. ДНК является записано в мРНК молекулы, которые перемещаются в рибосома где мРНК используется в качестве матрицы для построения белковой цепи. Поскольку нуклеиновые кислоты могут связываться с молекулами с дополнительный последовательности, существует различие между "смысл «последовательности, которые кодируют белки, и комплементарная« антисмысловая »последовательность, которая сама по себе нефункциональна, но может связываться со смысловой цепью.
Определение последовательности
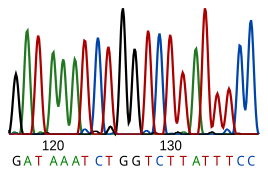
Секвенирование ДНК - это процесс определения нуклеотид последовательность данного ДНК фрагмент. Последовательность ДНК живого существа кодирует необходимую информацию для того, чтобы живое существо могло выжить и воспроизвести. Следовательно, определение последовательности полезно в фундаментальных исследованиях того, почему и как живут организмы, а также в прикладных предметах. Из-за важности ДНК для живых существ знание последовательности ДНК может быть полезно практически в любом биологическом исследование. Например, в лекарство его можно использовать для идентификации, диагностировать и потенциально развивать лечение за генетические заболевания. Точно так же исследование патогены может привести к лечению заразных заболеваний. Биотехнологии - это развивающаяся дисциплина, которая может предложить множество полезных продуктов и услуг.
РНК напрямую не секвенируется. Вместо этого он копируется в ДНК обратная транскриптаза, и эта ДНК затем секвенируется.
Современные методы секвенирования полагаются на дискриминационную способность ДНК-полимераз и, следовательно, могут различать только четыре основания. Инозин (образованный из аденозина во время Редактирование РНК ) читается как G, а 5-метилцитозин (образуется из цитозина посредством Метилирование ДНК ) читается как C. С современными технологиями трудно секвенировать небольшие количества ДНК, поскольку сигнал слишком слаб для измерения. Это преодолевается полимеразной цепной реакции (ПЦР) амплификация.
Цифровое представление

После получения последовательности нуклеиновой кислоты из организма она сохраняется. in silico в цифровом формате. Цифровые генетические последовательности могут храниться в базы данных последовательностей, быть проанализированы (см. Анализ последовательности ниже), могут быть изменены в цифровом виде и использоваться в качестве шаблонов для создания новой реальной ДНК с использованием искусственный синтез генов.
Анализ последовательности
Цифровые генетические последовательности могут быть проанализированы с использованием инструментов биоинформатика чтобы попытаться определить его функцию.
Генетическое тестирование
ДНК в организме геном можно проанализировать диагностировать уязвимости к унаследованным болезни, а также может использоваться для определения отцовства ребенка (генетический отец) или человека происхождение. Обычно каждый человек носит с собой по два варианта каждого ген, один унаследовал от матери, другой - от отца. В человеческий геном считается, что он содержит около 20 000–25 000 генов. Помимо учебы хромосомы на уровне отдельных генов генетическое тестирование в более широком смысле включает биохимический тесты на возможное наличие генетические заболевания, или мутантные формы генов, связанные с повышенным риском развития генетических нарушений.
Генетическое тестирование выявляет изменения в хромосомах, генах или белках.[6] Обычно тестирование используется для выявления изменений, связанных с наследственными заболеваниями. Результаты генетического теста могут подтвердить или исключить подозреваемое генетическое заболевание или помочь определить вероятность развития или передачи генетического заболевания у человека. В настоящее время используется несколько сотен генетических тестов, и еще больше разрабатываются.[7][8]
Выравнивание последовательности
В биоинформатике выравнивание последовательностей - это способ упорядочивания последовательностей ДНК, РНК, или же белок для выявления областей сходства, которые могут быть связаны с функциональными, структурный, или же эволюционный отношения между последовательностями.[9] Если две последовательности в выравнивании имеют общего предка, несоответствия можно интерпретировать как точечные мутации и пробелы как инсерционные или делеционные мутации (инделы ) введены в одну или обе линии за то время, когда они расходились друг от друга. При выравнивании последовательностей белков степень сходства между аминокислоты занятие определенной позиции в последовательности можно интерпретировать как грубую меру того, как консервированный конкретный регион или мотив последовательности находится среди родословных. Отсутствие замен или наличие только очень консервативных замен (то есть замена аминокислот, боковые цепи имеют аналогичные биохимические свойства) в определенной области последовательности, предполагают[10] что этот регион имеет структурное или функциональное значение. Хотя ДНК и РНК нуклеотид основания более похожи друг на друга, чем аминокислоты, сохранение пар оснований может указывать на аналогичную функциональную или структурную роль.[11]
Вычислительная филогенетика широко использует выравнивание последовательностей при построении и интерпретации филогенетические деревья, которые используются для классификации эволюционных взаимоотношений между гомологичными генами, представленными в геномах различных видов. Степень, в которой последовательности в наборе запроса различаются, качественно связана с эволюционным расстоянием последовательностей друг от друга. Грубо говоря, высокая идентичность последовательностей предполагает, что рассматриваемые последовательности имеют сравнительно молодой возраст. самый последний общий предок, в то время как низкая идентичность предполагает, что дивергенция более древняя. Это приближение, отражающее "молекулярные часы "гипотеза о том, что примерно постоянная скорость эволюционного изменения можно использовать для экстраполяции времени, прошедшего с момента первого расхождения двух генов (то есть слияние время), предполагает, что эффекты мутации и отбор постоянны по всем линиям последовательностей. Следовательно, он не учитывает возможные различия между организмами или видами в скорости Ремонт ДНК или возможная функциональная консервация конкретных областей в последовательности. (В случае нуклеотидных последовательностей гипотеза молекулярных часов в ее самой основной форме также не учитывает разницу в скорости принятия между тихие мутации которые не меняют значения данного кодон и другие мутации, которые приводят к другому аминокислота встраивается в белок.) Более статистически точные методы позволяют варьировать скорость эволюции на каждой ветви филогенетического дерева, тем самым обеспечивая более точные оценки времени слияния генов.
Последовательные мотивы
Часто первичная структура кодирует мотивы, которые имеют функциональное значение. Некоторые примеры мотивов последовательности: C / D[12]и коробки H / ACA[13]из snoRNAs, Sm сайт привязки обнаружены в сплайсосомных РНК, таких как U1, U2, U4, U5, U6, U12 и U3, то Последовательность Шайна-Далгарно,[14]в Консенсусная последовательность Козака[15]и Терминатор РНК-полимеразы III.[16]
Корреляции дальнего действия
Peng et al.[17][18] обнаружили существование дальних корреляций в некодирующих последовательностях пар оснований ДНК. Напротив, такие корреляции, похоже, не проявляются в кодирующих последовательностях ДНК. Это открытие было объяснено Grosberg et al.[19] глобальной пространственной структурой ДНК.
Энтропия последовательности
В Биоинформатика, энтропия последовательности, также известная как сложность последовательности или информационный профиль,[20] представляет собой числовую последовательность, обеспечивающую количественную меру локальной сложности последовательности ДНК, независимо от направления обработки. Манипуляции с информационными профилями позволяют анализировать последовательности с использованием методов без выравнивания, таких как, например, обнаружение мотивов и перестроек.[20][21][22]
Смотрите также
Рекомендации
- ^ а б Номенклатура не полностью определенных оснований в последовательностях нуклеиновых кислот, NC-IUB, 1984.
- ^ Номенклатурный комитет Международного союза биохимиков (NC-IUB) (1984). «Номенклатура неполностью определенных оснований в последовательностях нуклеиновых кислот». Получено 2008-02-04.
- ^ «BIOL2060: Перевод». mun.ca.
- ^ "Исследование". uw.edu.pl.
- ^ Nguyen, T; Брансон, Д.; Креспи, C L; Penman, BW; Wishnok, JS; Танненбаум, С. Р. (апрель 1992 г.). «Повреждение ДНК и мутации в клетках человека, подвергшихся действию оксида азота in vitro». Proc Natl Acad Sci USA. 89 (7): 3030–034. Bibcode:1992ПНАС ... 89.3030Н. Дои:10.1073 / пнас.89.7.3030. ЧВК 48797. PMID 1557408.
- ^ "Что такое генетическое тестирование?". Домашний справочник по генетике. 16 марта 2015.
- ^ «Генетическое тестирование». nih.gov.
- ^ «Определения генетического тестирования». Определения генетического тестирования (Хорхе Секейрос и Барбара Гимарайнш). EuroGentest Network of Excellence Project. 11 сентября 2008 г. Архивировано из оригинал 4 февраля 2009 г.. Получено 2008-08-10.
- ^ Крепление DM. (2004). Биоинформатика: анализ последовательности и генома (2-е изд.). Пресса лаборатории Колд-Спринг-Харбор: Колд-Спринг-Харбор, Нью-Йорк. ISBN 0-87969-608-7.
- ^ Ng, P.C .; Геникофф, С. (2001). "Прогнозирование вредных аминокислотных замен". Геномные исследования. 11 (5): 863–74. Дои:10.1101 / гр.176601. ЧВК 311071. PMID 11337480.
- ^ Витцани, Г. (2016). «Важнейшие шаги к жизни: от химических реакций до кода с использованием агентов». Биосистемы. 140: 49–57. Дои:10.1016 / j.biosystems.2015.12.007. PMID 26723230.
- ^ Самарский Д.А.; Фурнье MJ; Певец RH; Бертран Э (1998). «Мотив C / D snoRNA box направляет ядрышковое нацеливание, а также связывает синтез и локализацию snoRNA». Журнал EMBO. 17 (13): 3747–57. Дои:10.1093 / emboj / 17.13.3747. ЧВК 1170710. PMID 9649444.
- ^ Гано, Филипп; Кайзерг-Феррер, Мишель; Поцелуй, Тамаш (1 апреля 1997 г.). «Семейство малых ядрышковых РНК бокс-АСА определяется эволюционно консервативной вторичной структурой и повсеместными элементами последовательности, необходимыми для накопления РНК». Гены и развитие. 11 (7): 941–56. Дои:10.1101 / gad.11.7.941. PMID 9106664.
- ^ Шайн Дж, Далгарно Л. (1975). «Детерминант специфичности цистронов в бактериальных рибосомах». Природа. 254 (5495): 34–38. Bibcode:1975Натура.254 ... 34С. Дои:10.1038 / 254034a0. PMID 803646. S2CID 4162567.
- ^ Козак М. (октябрь 1987 г.). «Анализ 5'-некодирующих последовательностей из 699 матричных РНК позвоночных». Нуклеиновые кислоты Res. 15 (20): 8125–48. Дои:10.1093 / нар / 15.20.8125. ЧВК 306349. PMID 3313277.
- ^ Богенхаген Д.Ф., Браун Д.Д. (1981). «Нуклеотидные последовательности в ДНК Xenopus 5S, необходимые для терминации транскрипции». Клетка. 24 (1): 261–70. Дои:10.1016/0092-8674(81)90522-5. PMID 6263489. S2CID 9982829.
- ^ Peng, C.-K .; Булдырев, С. В .; Goldberger, A. L .; Havlin, S .; Sciortino, F .; Simons, M .; Стэнли, Х. Э. (1992). «Дальние корреляции в нуклеотидных последовательностях». Природа. 356 (6365): 168–70. Bibcode:1992Натура.356..168П. Дои:10.1038 / 356168a0. ISSN 0028-0836. PMID 1301010. S2CID 4334674.
- ^ Peng, C.-K .; Булдырев, С. В .; Havlin, S .; Simons, M .; Stanley, H.E .; Гольдбергер, А. Л. (1994). «Мозаичная организация нуклеотидов ДНК». Физический обзор E. 49 (2): 1685–89. Bibcode:1994PhRvE..49.1685P. Дои:10.1103 / PhysRevE.49.1685. ISSN 1063-651X. PMID 9961383.
- ^ Гросберг, А; Рабин, Y; Хэвлин, S; Нир, А (1993). «Смятая глобула, модель трехмерной структуры ДНК». Письма еврофизики. 23 (5): 373–78. Bibcode:1993EL ..... 23..373G. Дои:10.1209/0295-5075/23/5/012.
- ^ а б Пинхо, А; Гарсия, S; Пратас, Д; Феррейра, П. (21 ноября 2013 г.). "Последовательности ДНК с первого взгляда". PLOS ONE. 8 (11): e79922. Bibcode:2013PLoSO ... 879922P. Дои:10.1371 / journal.pone.0079922. ЧВК 3836782. PMID 24278218.
- ^ Пратас, Д; Silva, R; Пинхо, А; Феррейра, П. (18 мая 2015 г.). «Метод без выравнивания для поиска и визуализации перестроек между парами последовательностей ДНК». Научные отчеты. 5: 10203. Bibcode:2015НатСР ... 510203П. Дои:10.1038 / srep10203. ЧВК 4434998. PMID 25984837.
- ^ Троянская, О; Арбелл, О; Корен, Y; Ландау, G; Большой, А (2002). «Профили сложности последовательностей прокариотических геномных последовательностей: быстрый алгоритм для расчета лингвистической сложности». Биоинформатика. 18 (5): 679–88. Дои:10.1093 / биоинформатика / 18.5.679. PMID 12050064.