Транскриптом - Transcriptome
В транскриптом это набор всех РНК стенограммы, включая кодирование и некодирование, у человека или популяции клетки. Этот термин также может иногда использоваться для обозначения все РНК, или просто мРНК, в зависимости от конкретного эксперимента. Период, термин транскриптом Портманто слов стенограмма и геном; это связано с процессом производства транскриптов во время биологического процесса транскрипция.
Ранние этапы аннотаций транскриптомов начались с кДНК библиотеки, изданные в 1980-х гг. Впоследствии появление высокопроизводительной технологии привело к появлению более быстрых и эффективных способов получения данных о транскриптоме. Для изучения транскриптома используются два биологических метода, а именно: Микрочип ДНК, метод на основе гибридизации и РНК-последовательность, основанный на последовательностях подход.[1] RNA-seq является предпочтительным методом и является доминирующим техника транскриптомики с 2010-х гг. Транскриптомика одиночных клеток позволяет отслеживать изменения транскрипта во времени в отдельных ячейках.
Данные, полученные с помощью транскриптома, используются в исследованиях, чтобы получить представление о таких процессах, как клеточная дифференциация, канцерогенез, регулирование транскрипции и открытие биомаркеров среди прочего. Данные, полученные транскриптомом, также находит приложения в создании филогенетические отношения в процессе эволюции и в in vitro оплодотворение. Транскриптом тесно связан с другими -ome на основе биологических областей исследования; это дополняет протеом и метаболом и охватывает переводчик, экзом, мейоме и танатотранскриптом которые можно рассматривать как некоторые поля, изучающие определенные типы транскриптов РНК. Существует множество общедоступных баз данных транскриптомов.
Этимология и история
Слово транскриптом Портманто слов стенограмма и геном. Он появился вместе с другими неологизмы сформированный с использованием суффиксов -ome и -комикс для обозначения всех исследований, проводимых в масштабе всего генома в областях наук о жизни и технологий. Таким образом, транскриптом и транскриптомика были одними из первых слов, появившихся вместе с геномом и протеомом.[2] Первое исследование, представляющее случай коллекции кДНК библиотека для шелковая моль мРНК была опубликована в 1979 году.[3] Первое плодотворное исследование, в котором упоминается и исследуется транскриптом организма, было опубликовано в 1997 году и описало 60 633 транскрипта, выраженных в С. cerevisiae с помощью серийный анализ экспрессии генов (МУДРЕЦ).[4] С развитием высокопроизводительных технологий и биоинформатика и последующее увеличение вычислительной мощности, стало все более эффективным и простым для описания и анализа огромных объемов данных.[2] Попытки охарактеризовать транскриптом стали более заметными с появлением автоматизированного секвенирования ДНК в 1980-х годах.[5] В 90-е годы выраженный тег последовательности секвенирование использовалось для идентификации генов и их фрагментов.[6] За этим последовали такие методы, как серийный анализ экспрессии генов (SAGE), кэп-анализ экспрессии генов (CAGE) и массовая параллельная последовательность подписей (МПСС).
Транскрипция
Транскриптом включает в себя все рибонуклеиновая кислота (РНК) транскрипты, присутствующие в данном организме или экспериментальном образце.[7] РНК - основной носитель генетической информации, отвечающий за процесс преобразования ДНК в фенотип организма. Ген может давать одноцепочечный информационная РНК (мРНК) посредством молекулярного процесса, известного как транскрипция; эта мРНК комплементарна цепи ДНК, из которой она произошла.[5] Фермент РНК-полимераза II прикрепляется к матричной цепи ДНК и катализирует добавление рибонуклеотиды к 3'-концу растущей последовательности транскрипта мРНК.[8]
Чтобы запустить свою функцию, РНК-полимераза II должна распознавать промоторная последовательность, расположенный выше (5 ') гена. У эукариот этот процесс опосредуется факторы транскрипции, в первую очередь Фактор транскрипции II D (TFIID), который распознает Коробка ТАТА и способствует позиционированию РНК-полимеразы в соответствующем стартовом сайте. Чтобы закончить производство транскрипта РНК, прекращение обычно происходит на расстоянии нескольких сотен нуклеотидов от терминирующей последовательности, и происходит расщепление.[8] Этот процесс происходит в ядре клетки вместе с Обработка РНК с помощью которых молекулы мРНК закрытый, сращенный и полиаденилированный для повышения их стабильности перед последующим попаданием в цитоплазму. МРНК дает белки в процессе перевод что происходит в рибосомы.
Типы транскриптов РНК
В соответствии с центральная догма молекулярной биологии, транскриптом изначально охватывал только транскрипты мРНК, кодирующие белок. Тем не менее существует несколько подтипов РНК с различными функциями. Многие транскрипты РНК не кодируют белок или выполняют различные регуляторные функции в процессе транскрипции и трансляции генов. Типы РНК, не подпадающие под действие центральная догма молекулярной биологии находятся некодирующие РНК которые можно разделить на две группы длинная некодирующая РНК и короткая некодирующая РНК.
Длинная некодирующая РНК включает все транскрипты некодирующей РНК, длина которых превышает 200 нуклеотидов. Члены этой группы составляют наибольшую часть некодирующего транскриптома. Короткая некодирующая РНК включает следующих членов:
- переносить РНК (тРНК)
- микро РНК (миРНК): длина 19-24 нуклеотида (нуклеотидов). Микро РНК повышают или понижают уровни экспрессии мРНК в процессе РНК-интерференция на посттранскрипционном уровне.[2]
- малая интерферирующая РНК (миРНК): 20-24 н.
- малая ядрышковая РНК (мяРНК)
- Piwi-взаимодействующая РНК (пиРНК): 24-31 н. Они взаимодействуют с Пиви белки из Аргонавт семьи и имеют функцию нацеливания и расщепления транспозоны.[9]
- энхансерная РНК (эРНК)[2]
Объем исследования
В геноме человека около 5% всех генов транскрибируются в РНК.[7] Транскриптом состоит из кодирующей мРНК, которая составляет около 1-4% от ее полноты, и некодирующих РНК, которые составляют остальную часть генома и не образуют белков.[10][11] Число последовательностей, не кодирующих белок, увеличивается у более сложных организмов.[12]
Некоторые факторы затрудняют установление содержания транскриптома. К ним относятся альтернативное сращивание, Редактирование РНК и альтернативная транскрипция среди прочего.[12] Кроме того, методы транскриптома способны улавливать транскрипцию, происходящую в образце в определенный момент времени, хотя содержание транскриптома может изменяться во время дифференцировки.[5] Основными целями транскриптомики являются следующие: «каталогизация всех видов транскриптов, включая мРНК, некодирующие РНК и малые РНК; определение структуры транскрипции генов с точки зрения их стартовых сайтов, 5 'и 3' концов, сплайсинг паттерны и другие посттранскрипционные модификации, а также для количественной оценки изменения уровней экспрессии каждого транскрипта во время развития и в различных условиях ».[1]
Термин может применяться к общему набору стенограмм в данном организм или к определенному подмножеству транскриптов, присутствующих в конкретном типе клеток. в отличие от геном, который примерно фиксируется для данной клеточной линии (исключая мутации ), транскриптом может меняться в зависимости от внешних условий окружающей среды. Поскольку он включает в себя все транскрипты мРНК в клетке, транскриптом отражает гены которые активно выразил в любой момент времени, за исключением явлений деградации мРНК, таких как ослабление транскрипции. Изучение транскриптомика, (который включает профилирование выражений, анализ вариантов стыковки и т.д.), исследует уровень экспрессии РНК в данной популяции клеток, часто фокусируясь на мРНК, но иногда включая другие, такие как тРНК и мРНК.
Способы строительства
Транскриптомика - это количественная наука, которая включает в себя назначение списка строк («прочтений») объекту («транскриптов» в геноме). Для расчета силы экспрессии подсчитывается плотность считываний, соответствующих каждому объекту.[13] Первоначально транскриптомы анализировались и изучались с помощью выраженные теги последовательности библиотеки и серийный и кэп-анализ экспрессии генов (SAGE).
В настоящее время два основных методы транскриптомики включают ДНК-микрочипы и РНК-Seq. Оба метода требуют выделения РНК через Извлечение РНК методы с последующим его отделением от других клеточных компонентов и обогащением мРНК.[14][15]
Существует два основных метода определения последовательностей транскриптомов. Один подход отображает последовательность считывания на эталонный геном либо самого организма (чей транскриптом изучается), либо близкородственного вида. Другой подход, de novo сборка транскриптома, использует программное обеспечение для вывода транскриптов непосредственно из считывания коротких последовательностей и используется в организмах с геномами, которые не секвенированы.[16]
ДНК-микрочипы

Первые исследования транскриптомов были основаны на микрочип техники (также известные как ДНК-чипы). Микроматрицы состоят из тонких слоев стекла с пятнами, на которых олигонуклеотиды, известные как «зонды» расположены в виде массива; каждое пятно содержит известную последовательность ДНК.[17]
При выполнении микроматричного анализа мРНК собирают из контрольного и экспериментального образцов, последний обычно представляет заболевание. Интересующая РНК превращается в кДНК для повышения ее стабильности и маркируется флуорофоры двух цветов, обычно зеленого и красного, для двух групп. КДНК наносят на поверхность микрочипа, где она гибридизуется с олигонуклеотидами на чипе, и для сканирования используется лазер. Интенсивность флуоресценции на каждом пятне микроматрицы соответствует уровню экспрессии гена, и на основании цвета выбранных флуорофоров можно определить, какой из образцов демонстрирует более высокие уровни интересующей мРНК.[6]
Один микрочип обычно содержит достаточно олигонуклеотидов для представления всех известных генов; однако данные, полученные с помощью микрочипов, не дают информации о неизвестных генах. В течение 2010-х годов микроматрицы были почти полностью заменены методами следующего поколения, основанными на секвенировании ДНК.
Секвенирование РНК
Секвенирование РНК - это секвенирование следующего поколения технологии; как таковой требуется только небольшое количество РНК и никаких предварительных знаний о геноме.[2] Он позволяет проводить как качественный, так и количественный анализ транскриптов РНК, первый из которых позволяет обнаруживать новые транскрипты, а второй позволяет измерить относительные количества транскриптов в образце.[9]
Три основных этапа секвенирования транскриптомов любых биологических образцов включают очистку РНК, синтез библиотеки РНК или кДНК и секвенирование библиотеки.[9] Процесс очистки РНК отличается для коротких и длинных РНК.[9] За этим шагом обычно следует оценка качества РНК с целью исключения таких загрязнений, как ДНК, или технических загрязнений, связанных с обработкой образцов. Качество РНК измеряется с помощью УФ-спектрометрии с пиком поглощения 260 нм.[18] Целостность РНК также можно проанализировать количественно, сравнивая соотношение и интенсивность 28S РНК к 18S РНК сообщается в счетчике числа целостности РНК (RIN).[18] Поскольку мРНК представляет собой интересующий вид и составляет только 3% от ее общего содержания, образец РНК следует обработать для удаления рРНК и тРНК и тканеспецифичных транскриптов РНК.[18]
Этап подготовки библиотеки с целью получения коротких фрагментов кДНК начинается с фрагментации РНК до транскриптов длиной от 50 до 300. пар оснований. Фрагментация может быть ферментативной (РНК эндонуклеазы ), химический (буфер тризмагниевой соли, химический гидролиз ) или механический (обработка ультразвуком, распыление).[19] Обратная транскрипция используется для преобразования матриц РНК в кДНК, и для этого можно использовать три метода прайминга, включая oligo-DT, с использованием случайных праймеров или лигирования специальных адаптерных олигонуклеотидов.
Транскриптомика одиночных клеток
Транскрипцию также можно изучать на уровне отдельных клеток с помощью одноклеточная транскриптомика. Секвенирование одноклеточной РНК (scRNA-seq) - это недавно разработанный метод, который позволяет анализировать транскриптом отдельных клеток. При одноклеточной транскриптомике также принимаются во внимание субпопуляции типов клеток, которые составляют интересующую ткань.[20] Этот подход позволяет определить, вызваны ли изменения в экспериментальных образцах фенотипическими клеточными изменениями, а не пролиферацией, при которой определенный тип клеток может быть сверхэкспрессирован в образце.[21] Кроме того, при оценке клеточного развития через дифференциация профили средней экспрессии могут упорядочивать клетки только по времени, а не по стадиям развития, и, следовательно, не могут показать тенденции в уровнях экспрессии генов, специфичные для определенных стадий.[22] Одноклеточные транскриптомные методы были использованы для характеристики редких популяций клеток, таких как циркулирующие опухолевые клетки, раковые стволовые клетки в солидных опухолях и эмбриональные стволовые клетки (ESC) у млекопитающих бластоцисты.[23]
Хотя стандартизированных методов транскриптомики одиночных клеток не существует, необходимо предпринять несколько шагов. Первый шаг включает в себя изоляцию ячеек, которую можно выполнить с использованием методов с низкой и высокой пропускной способностью. За этим следует стадия кПЦР, а затем одноклеточная RNAseq, на которой интересующая РНК превращается в кДНК. Новейшие разработки в одноклеточной транскриптомике позволяют сохранять тканевую и субклеточную локализацию посредством криосрезов тонких срезов тканей и секвенирования транскриптома в каждом срезе. Другой метод позволяет визуализировать отдельные транскрипты под микроскопом, сохраняя при этом пространственную информацию каждой отдельной клетки, в которой они экспрессируются.[23]
Анализ
Был создан и аннотирован ряд баз данных транскриптомов, специфичных для организмов, чтобы помочь в идентификации генов, которые по-разному экспрессируются в разных популяциях клеток.
РНК-последовательность (2013 г.) как предпочтительный метод измерения транскриптомов организмов, хотя более старый метод измерения ДНК-микрочипы все еще используется.[1] RNA-seq измеряет транскрипцию определенного гена путем преобразования длинных РНК в библиотеку кДНК фрагменты. Затем фрагменты кДНК секвенируют с использованием технологии высокопроизводительного секвенирования и выравнивают по эталонному геному или транскриптому, который затем используется для создания профиля экспрессии генов.[1]
Приложения
Млекопитающие
Стенограммы стволовые клетки и рак клетки представляют особый интерес для исследователей, которые стремятся понять процессы клеточная дифференциация и канцерогенез. Конвейер с использованием данных RNA-seq или массива генов может использоваться для отслеживания генетических изменений, происходящих в корень и клетки-предшественники и требует, по крайней мере, трех независимых данных об экспрессии генов от первого типа клеток и зрелых клеток.[24]
Анализ транскриптомов человека ооциты и эмбрионы используется для понимания молекулярных механизмов и сигнальных путей, контролирующих раннее эмбриональное развитие, и теоретически может быть мощным инструментом для правильного отбор эмбрионов в экстракорпоральное оплодотворение.[нужна цитата ] Анализы содержания транскриптомов плаценты в I триместре беременности в in vitro оплодотворение и перенос эмбрионов (IVT-ET) выявили различия в генетической экспрессии, которые связаны с более высокой частотой неблагоприятных перинатальных исходов. Такое понимание можно использовать для оптимизации практики.[25] Анализ транскриптома также может использоваться для оптимизации криоконсервации ооцитов за счет снижения травм, связанных с этим процессом.[26]
Транскриптомика - это новая и постоянно развивающаяся область в биомаркер открытие для использования при оценке безопасности лекарств или химических оценка рисков.[27]
Транскриптомы также могут использоваться для вывести филогенетические отношения среди людей или для выявления эволюционных паттернов сохранения транскриптома[28].
Анализ транскриптомов был использован для выявления случаев антисмысловой транскрипции, их роли в экспрессии генов через взаимодействие с окружающими генами и их количества в различных хромосомах.[29] RNA-seq также использовался, чтобы показать, как изоформы РНК, транскрипты, происходящие от одного и того же гена, но с разными структурами, могут производить сложные фенотипы из ограниченных геномов.[16]
Растения
Анализ транскриптома был использован для изучения эволюция и процесс диверсификации видов растений. В 2014 г. Проект "1000 геномов растений" был завершен, в котором транскриптомы 1124 видов растений из семейств viridiplantae, глаукофита и родофита были секвенированы. Последовательности, кодирующие белок, впоследствии сравнивали, чтобы сделать вывод о филогенетических отношениях между растениями и охарактеризовать время их появления. диверсификация в процессе эволюции.[30] Исследования транскриптома использовались для характеристики и количественной оценки экспрессии генов в зрелых пыльца. Было обнаружено, что гены, участвующие в метаболизме клеточной стенки и цитоскелета, сверхэкспрессируются. Транскриптомные подходы также позволяют отслеживать изменения в экспрессии генов на разных стадиях развития пыльцы, от микроспор до зрелых пыльцевых зерен; кроме того, такие стадии можно было сравнивать между видами разных растений, включая Арабидопсис, рис и табак.[31]
Отношение к другим областям омэ
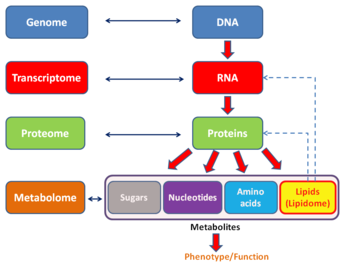
Подобно другим -ome основанные на технологиях, анализ транскриптома позволяет беспристрастно подходить к проверке гипотез экспериментально. Этот подход также позволяет открывать новые медиаторы в сигнальных путях.[13] Как и в случае с другими технологиями, основанными на -omics, транскриптом может быть проанализирован в рамках мультиомика подход. Это дополняет метаболомика но вопреки протеомике прямая связь между транскриптом и метаболит не может быть установлено.
Есть несколько полей -ome, которые можно рассматривать как подкатегории транскриптома. В экзом отличается от транскриптома тем, что он включает только те молекулы РНК, которые встречаются в определенной популяции клеток, и обычно включает количество или концентрацию каждой молекулы РНК в дополнение к молекулярным идентичностям. Кроме того, транскритпом также отличается от переводчик, который представляет собой набор РНК, подвергающихся трансляции.
Термин мейом используется в функциональная геномика для описания мейотического транскриптома или набора транскриптов РНК, полученных в процессе мейоз.[32] Мейоз - ключевая особенность репродуктивного здоровья половым путем. эукариоты, и включает в себя соединение гомологичная хромосома, синапс и рекомбинация. Поскольку мейоз у большинства организмов происходит за короткий период времени, профилирование мейотических транскриптов затруднено из-за проблемы выделения (или обогащения) мейотических клеток (мейоциты ). Как и в случае анализа транскриптома, мейом можно изучать на уровне всего генома с использованием крупномасштабных транскриптомных методов.[33] Мейом хорошо охарактеризован у млекопитающих и дрожжевых систем и несколько менее широко охарактеризован у растений.[34]
В танатотранскриптом состоит из всех транскриптов РНК, которые продолжают экспрессироваться или начинают повторно экспрессироваться во внутренних органах мертвого тела через 24-48 часов после смерти. Некоторые гены включают те, которые подавляются после развитие плода. Если танатотранскриптом связан с процессом запрограммированной гибели клеток (апоптоз ), его можно назвать апоптотическим танатотранскриптомом. Анализы танатотранскриптома используются в судебная медицина.[35]
eQTL картирование можно использовать для дополнения геномики транскриптомикой; генетические варианты на уровне ДНК, а экспрессия генов измеряется на уровне РНК.[36]
Отношение к протеому
Транскриптом можно рассматривать как подмножество протеом, то есть весь набор белков, экспрессируемых геномом.
Однако анализ относительных уровней экспрессии мРНК может быть затруднен тем фактом, что относительно небольшие изменения в экспрессии мРНК могут вызывать большие изменения общего количества соответствующего белка, присутствующего в клетке. Один метод анализа, известный как анализ обогащения набора генов, идентифицирует корегулируемые генные сети, а не отдельные гены, которые активируются или подавляются в разных популяциях клеток.[1]
Хотя исследования микроматрицы могут выявить относительные количества различных мРНК в клетке, уровни мРНК не прямо пропорциональны уровню экспрессии белки они кодируют.[37] Количество белковых молекул, синтезируемых с использованием данной молекулы мРНК в качестве матрицы, в значительной степени зависит от особенностей инициации трансляции последовательности мРНК; в частности, способность последовательности инициации трансляции является ключевым фактором при привлечении рибосомы для белка перевод.
Базы данных транскриптомов
Смотрите также
- Технологии транскриптомики
- Серийный анализ экспрессии генов
- Список тем омиков по биологии
- Метаболом
- Экспрессия гена
- Сетевой анализ взвешенной коэкспрессии генов
- Функциональная геномика
Примечания
- ^ а б c d Ван, Чжун; Герштейн, Марк; Снайдер, Майкл (январь 2009 г.). «RNA-Seq: революционный инструмент для транскриптомики». Природа Обзоры Генетика. 10 (1): 57–63. Дои:10.1038 / nrg2484. ЧВК 2949280. PMID 19015660.
- ^ а б c d е Хименес-Чилларон, Josep C .; Диас, Рубен; Рамон-Крауэль, Марта (2014). «Глава 4 - Инструменты Omics для полногеномного анализа метилирования и модификаций гистонов». Комплексная аналитическая химия. 64: 81–110. Дои:10.1016 / B978-0-444-62651-6.00004-0. Получено 25 апреля 2020.
- ^ ГК, Сим; ФК, Кафатос; CW, Джонс; Доктор медицины, Келер; А, Эфстратиадис; Т., Маниатис (декабрь 1979 г.). «Использование библиотеки кДНК для изучения эволюции и развития экспрессии мультигенных семейств хориона». Клетка. 8 (4): 1303–16. Дои:10.1016/0092-8674(79)90241-1. PMID 519770.
- ^ Е. Велкулеску, Виктор; Чжан, Линь; Чжоу, Вэй; Фогельштейн, Якоб; Басрай, Мунира; Э. Бассет-младший, Дуглас; Хитер, Фил; Фогельштейн, Берт; В. Кинзлер, Кеннет (1997). «Характеристика дрожжевого транскриптома». Клетка. 2 (88): 243–51. Дои:10.1016 / S0092-8674 (00) 81845-0. PMID 9008165. S2CID 11430660.
- ^ а б c Перальта, Михаэла (2012). "Человеческий транскриптом: незаконченная история". Гены. 3 (3): 344–360. Дои:10.3390 / гены3030344. ЧВК 3422666. PMID 22916334.
- ^ а б Говиндараджан, Раджешвар; Дурайян, Джеяпрадха; Калиаппан, Карунакаран; Паланисами, Муругесан (2012). «Микромассив и его приложения». Журнал фармации и биологических наук. 4 (6): S310-2. Дои:10.4103/0975-7406.100283. ЧВК 3467903. PMID 23066278.
- ^ а б К. Фрит, Мартин; Фазан, Майкл; С. Маттик, Джон (2005). «Геномика: удивительная сложность человеческого транскриптома». Европейский журнал генетики человека. 13 (8): 894–897. Дои:10.1038 / sj.ejhg.5201459. PMID 15970949. S2CID 2836126.
- ^ а б Клэнси, Сюзанна (2008). «Транскрипция ДНК». Природное образование. 1 (11): 41.
- ^ а б c d Cellerino & Sanguanini 2018, п. 12
- ^ Берг JMTJ, Страйер Л. Биохимия. Нью-Йорк: У. Фриман, 2002
- ^ Маттик Дж.С., Макунин И.В. Некодирующая РНК. Hum Mol Genet 2006; 15 Spec № 1: R17–29
- ^ а б У. Адамс, Джилл (2008). «Транскриптом: связь генома с функцией генов». Природное образование. 1 (1): 195.
- ^ а б Cellerino & Sanguanini 2018, п. предисловие
- ^ Брайант С., Мэннинг Д.Л. (1998). «Выделение информационной РНК». Протоколы выделения и характеристики РНК. Методы молекулярной биологии. 86. С. 61–4. Дои:10.1385/0-89603-494-1:61. ISBN 978-0-89603-494-5. PMID 9664454.
- ^ Хомчинский П., Сакки Н. (апрель 1987 г.). «Одностадийный метод выделения РНК кислотной экстракцией тиоцианат-фенол-хлороформ гуанидиния». Аналитическая биохимия. 162 (1): 156–9. Дои:10.1016/0003-2697(87)90021-2. PMID 2440339.
- ^ а б Тачибана, Крис (31 июля 2015 г.). «Транскриптомика сегодня: микроматрицы, последовательность РНК и многое другое».. Научный журнал. 349 (6247): 544. Bibcode:2015Научный ... 349..544Т. Получено 2 мая 2020.
- ^ Schena, M .; Shalon, D .; Дэвис, Р. У .; Браун, П. О. (20 октября 1995 г.). «Количественный мониторинг паттернов экспрессии генов с помощью комплементарного ДНК-микрочипа». Наука. Нью-Йорк, штат Нью-Йорк). 270 (5235): 467–470. Bibcode:1995Научный ... 270..467S. Дои:10.1126 / science.270.5235.467. ISSN 0036-8075. PMID 7569999. S2CID 6720459.
- ^ а б c Cellerino & Sanguanini 2018, п. 13
- ^ Cellerino & Sanguanini 2018, п. 18
- ^ Кантер, Итамар; Калиский, Томер (10 марта 2015 г.). «Транскриптомика отдельной клетки: методы и приложения». Границы онкологии. 5: 53. Дои:10.3389 / fonc.2015.00053. ISSN 2234-943X. ЧВК 4354386. PMID 25806353.
- ^ Стегл, Оливер; А. Тайхманн, Сара; К. Мариони, Джон (2015). «Вычислительные и аналитические проблемы в одноклеточной транскриптомике». Природа Обзоры Генетика. 16 (3): 133–45. Дои:10.1038 / nrg3833. PMID 25628217. S2CID 205486032.
- ^ Трапнелл, Коул (1 октября 2015 г.). «Определение типов и состояний клеток с помощью одноклеточной геномики». Геномные исследования. 25 (10): 1491–1498. Дои:10.1101 / гр.190595.115. ISSN 1088-9051. ЧВК 4579334. PMID 26430159.
- ^ а б Кантер, Итамар; Калиский, Томер (2015). «Транскриптомика одиночных клеток: методы и приложения». Границы онкологии. 5 (13). Дои:10.3389 / fonc.2015.00053. ЧВК 4354386. PMID 25806353.
- ^ Годой, Патрисио; Шмидт-Хек, Вольфганг; Хеллвиг, Бирте; Нелл, Патрик; Фейерборн, Дэвид; Раненфюрер Йорг; Каттлер, Катрин; Вальтер, Йорн; Блютген, Нильс; Г. Хенгстлер, янв (5 июля 2018 г.). «Оценка дифференцировки стволовых клеток на основе полногеномных профилей экспрессии». Философские труды Королевского общества B. 373 (1750): 20170221. Дои:10.1098 / rstb.2017.0221. ЧВК 5974444. PMID 29786556.
- ^ Чжао, L; Чжэн, X; Лю, Дж; Zheng, R; Ян, Р; Ван, Y; Вс, Л. (1 июля 2019 г.). «На транскриптом плаценты в первом триместре плаценты влияет оплодотворение in vitro и перенос эмбриона». Репродуктивная биология и эндокринология. 17 (1): 50. Дои:10.1186 / s12958-019-0494-7. ЧВК 6604150. PMID 31262321.
- ^ Эроглу, Биннур; А. Шурек, Эдита; Шалл, Питер; Э. Лэтэм, Кейт; Эроглу, Али (6 апреля 2020 г.). «Исследование стойких криоповреждений транскриптома ооцит-эмбрион». PLOS ONE. 15 (4): e0231108. Bibcode:2020PLoSO..1531108E. Дои:10.1371 / journal.pone.0231108. ЧВК 7135251. PMID 32251418.
- ^ Сабо, Давид (2014). «Транскриптомные биомаркеры в оценке безопасности и риска химических веществ». Транскриптомные биомаркеры в оценке безопасности и риска химических веществ. В Рамеш Гупта, редакторы: Гупта - Биомаркеры в токсикологии, Оксфорд: Academic Press. С. 1033–1038. Дои:10.1016 / B978-0-12-404630-6.00062-2. ISBN 978-0-12-404630-6.
- ^ Дрост, Хайк-Георг; Габель, Александр; Гроссе, Иво; Квинт, Марсель; Гросс, Иво (2018-05-01). «myTAI: эволюционная транскриптомика с R». Биоинформатика. 34 (9): 1589–1590. Дои:10.1093 / molbev / msv012. ISSN 0737-4038. ЧВК 5925770. PMID 29309527.
- ^ S, Катаяма; и другие. (2005). «Антисмысловая транскрипция в транскриптоме млекопитающих». Наука. 309 (5740): 1564–6. Bibcode:2005Научный ... 309.1564R. Дои:10.1126 / science.1112009. PMID 16141073. S2CID 34559885.
- ^ Инициатива "Тысяча расшифровок растений" (23 октября 2019 г.). «Тысяча транскриптомов растений и филогеномика зеленых растений». Природа. 574 (7780): 679–685. Дои:10.1038 / s41586-019-1693-2. ЧВК 6872490. PMID 31645766.
- ^ Ратли, Николас; Твелл, Дэвид (12 марта 2015 г.). «Десятилетие транскриптомики пыльцы». Размножение растений. 28 (2): 73–89. Дои:10.1007 / s00497-015-0261-7. ЧВК 4432081. PMID 25761645.CS1 maint: дата и год (ссылка на сайт)
- ^ Крисмани, Уэйн; Бауманн, Юте; Саттон, Тим; Ширли, Нил; Вебстер, Трейси; Спангенберг, немецкий; Лэнгридж, Питер; Способный, Джейсон (2006). «Микроматричный анализ экспрессии мейоза и микроспорогенеза в гексаплоидной мягкой пшенице». BMC Genomics. 7 (267): 267. Дои:10.1186/1471-2164-7-267. ЧВК 1647286. PMID 17052357.
- ^ Д. Бовилл, Уильям; Девешвар, Приянка; Капур, Санджай; А. Эйбл, Джейсон (2009). «Полногеномные подходы для выявления кандидатов на ранние мейотические гены в зерновых». Функциональная и интегративная геномика. 9 (2): 219–29. Дои:10.1007 / s10142-008-0097-4. PMID 18836753. S2CID 22854431.
- ^ Девешвар, Приянка; Д. Бовилл, Уильям; Шарма, Рита; Able, Джейсон; Капур, Санджай (9 мая 2011 г.). «Анализ транскриптомов пыльников для выявления генов, способствующих развитию мейоза и мужских гаметофитов у риса». BMC Биология растений. 11 (78): 78. Дои:10.1186/1471-2229-11-78. ЧВК 3112077. PMID 21554676.
- ^ Javan, G.T .; Могу я.; Финли, С. Дж .; Сони, S (2015). «Апоптотический танатотранскриптом, связанный с печенью трупов». Судебная медицина, медицина и патология. 11 (4): 509–516. Дои:10.1007 / s12024-015-9704-6. PMID 26318598. S2CID 21583165.
- ^ Мандзони, Клаудиа; Киа, Демис; Вандровцова, Яна; Харди, Джон; Вуд, Николас; Льюис, Патрик; Феррари, Раффаэле (март 2018 г.). «Геном, транскриптом и протеом: распространение данных omics и их интеграция в биомедицинские науки». Брифинги по биоинформатике. 19 (2): 286–302. Дои:10.1093 / нагрудник / bbw114. ЧВК 6018996. PMID 27881428.
- ^ Шванхойссер, Бьёрн; и другие. (Май 2011 г.). «Глобальная количественная оценка контроля экспрессии генов млекопитающих» (PDF). Природа. 473 (7347): 337–342. Bibcode:2011Натура.473..337S. Дои:10.1038 / природа10098. PMID 21593866. S2CID 205224972.
Рекомендации
- Селлерино, А; Сангуанини, М. (2018), Анализ транскриптома: введение и примеры из неврологии, Дои:10.1007/978-88-7642-642-1, ISBN 978-88-7642-641-4
дальнейшее чтение
- ^ Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. (2005). Анализ обогащения набора генов: основанный на знаниях подход для интерпретации профилей экспрессии в масштабе всего генома. Proc Natl Acad Sci USA 102(43):15545-50.
- ^ Лаул О., Хирш-Хоффманн М., Хруз Т., Груиссем В. и П. Циммерманн. (2006) Интернет-анализ транскриптома мыши с помощью Genevestigator. BMC Bioinformatics 7:311
- ^ Assou, S .; Boumela, I .; Haouzi, D .; Анахори, Т .; Dechaud, H .; De Vos, J .; Хамама, С. (2010). «Динамические изменения в экспрессии генов во время раннего развития человеческого эмбриона: от фундаментальных аспектов до клинического применения». Обновление репродукции человека. 17 (2): 272–290. Дои:10.1093 / humupd / dmq036. ЧВК 3189516. PMID 20716614.
- ^ Огородников А; Каргаполова, Ы; Данквардт, С. (2016). «Процессинг и расширение транскриптома на 3'-конце мРНК при здоровье и болезни: поиск правильного конца». Eur J Physiol. 468 (6): 993–1012. Дои:10.1007 / s00424-016-1828-3. ЧВК 4893057. PMID 27220521.
| Геномика | |
|---|---|
| Биоинформатика | |
| Структурная биология | |
| Инструменты исследования | |
| Организации |
|
| |