Геномика - Genomics
| Часть набор на |
| Генетика |
|---|
 |
| Ключевые компоненты |
| История и темы |
| Исследование |
| Персонализированная медицина |
| Персонализированная медицина |
Геномика это междисциплинарная область биология уделяя особое внимание структуре, функциям, развитию, отображению и редактированию геномы. Геном - это полный набор организмов ДНК, включая все его гены. В отличие от генетика, который относится к изучению физическое лицо генов и их роли в наследовании, геномика направлена на коллективную характеристику и количественную оценку все генов организма, их взаимосвязь и влияние на организм.[1] Гены могут управлять производством белки с помощью ферментов и молекул-посредников. В свою очередь, белки составляют структуры тела, такие как органы и ткани, а также контролируют химические реакции и передают сигналы между клетками. Геномика также включает в себя секвенирование и анализ геномов с использованием высокой производительности. Секвенирование ДНК и биоинформатика собирать и анализировать функции и структуру целых геномов.[2][3][4] Достижения в области геномики вызвали революцию в исследованиях, основанных на открытиях, и системная биология чтобы облегчить понимание даже самых сложных биологических систем, таких как мозг.[5]
Эта область также включает исследования внутригеномных (внутри генома) явлений, таких как эпистаз (влияние одного гена на другой), плейотропия (один ген влияет на более чем одну черту), гетерозис (сила гибрида) и другие взаимодействия между места и аллели внутри генома.[6]
История
Этимология
От греческого ΓΕΝ[7] ген, «ген» (гамма, эпсилон, ню, эпсилон), означающий «становиться, создавать, сотворение, рождение», и последующие варианты: генеалогия, генезис, генетика, генный, геномер, генотип, род и т. д. геном (от Немецкий Геном, приписываемый Ханс Винклер ) использовался в английский еще в 1926 г.[8] период, термин геномика был придуман Томом Родериком, генетик на Лаборатория Джексона (Бар-Харбор, Мэн ), за пивом на встрече в Мэриленд о картировании генома человека в 1986 году.[9]
Усилия по раннему секвенированию
Следующий Розалинд Франклин подтверждение спиральной структуры ДНК, Джеймс Д. Уотсон и Фрэнсис Крик публикация структуры ДНК в 1953 г. и Фред Сэнгер публикация Аминокислота последовательности инсулина в 1955 г., секвенирование нуклеиновых кислот стало основной целью ранних молекулярные биологи.[10] В 1964 г. Роберт В. Холли и его коллеги опубликовали первую когда-либо определенную последовательность нуклеиновой кислоты, рибонуклеотид последовательность аланин переносить РНК.[11][12] Продлевая эту работу, Маршалл Ниренберг и Филип Ледер раскрыл триплетный характер генетический код и смогли определить последовательности 54 из 64 кодоны в своих экспериментах.[13] В 1972 г. Уолтер Фирс и его команда в лаборатории молекулярной биологии Гентский университет (Гент, Бельгия ) первыми определили последовательность гена: гена для Бактериофаг MS2 белок оболочки.[14] Группа Файерса расширила свою работу с белками оболочки MS2, определив полную нуклеотидную последовательность MS2-РНК бактериофага (чей геном кодирует всего четыре гена из 3569 пар оснований [bp]) и Обезьяний вирус 40 в 1976 и 1978 годах соответственно.[15][16]
Разработана технология секвенирования ДНК


Помимо своей основополагающей работы по аминокислотной последовательности инсулина, Фредерик Сэнгер и его коллеги сыграли ключевую роль в разработке методов секвенирования ДНК, которые позволили создать комплексные проекты секвенирования генома.[6] В 1975 году он и Алан Коулсон опубликовали процедуру секвенирования с использованием ДНК-полимеразы с радиоактивно меченными нуклеотидами, которую он назвал Техника плюс и минус.[17][18] Это включало два тесно связанных метода, которые генерировали короткие олигонуклеотиды с определенными 3'-концами. Их можно разделить на электрофорез на полиакриламид гель (так называемый электрофорез в полиакриламидном геле) и визуализировали с помощью авторадиографии. Эта процедура позволяла секвенировать до 80 нуклеотидов за один раз и была большим улучшением, но все же была очень трудоемкой. Тем не менее в 1977 году его группе удалось секвенировать большую часть из 5386 нуклеотидов одноцепочечной бактериофаг φX174, завершая первый полностью секвенированный геном на основе ДНК.[19] Утонченность Плюс и Минус метод привел к обрыву цепи, или Метод Сенгера (увидеть ниже ), которые легли в основу методов секвенирования ДНК, картирования генома, хранения данных и биоинформатического анализа, наиболее широко используемых в последующие четверть века исследований.[20][21] В том же году Уолтер Гилберт и Аллан Максам из Гарвардский университет независимо разработал Максам-Гилберт метод (также известный как химический метод) секвенирования ДНК, предполагающего преимущественное расщепление ДНК по известным основаниям, менее эффективный метод.[22][23] За свою новаторскую работу по секвенированию нуклеиновых кислот Гилберт и Сэнджер разделили половину 1980 года. Нобелевская премия по химии с Пол Берг (рекомбинантная ДНК ).
Полные геномы
Появление этих технологий привело к быстрому увеличению масштабов и скорости выполнения проекты секвенирования генома. Первая полная последовательность генома эукариотическая органелла, человек митохондрия (16,568 п.н., около 16,6 т.п.н. [килобаза]), было сообщено в 1981 г.,[24] и первый хлоропласт геномы последовали в 1986 году.[25][26] В 1992 г. первый эукариот хромосома, хромосома III пивных дрожжей Saccharomyces cerevisiae (315 т.п.н.) секвенировали.[27] Первый свободноживущий организм, который был подвергнут секвенированию, был Haemophilus influenzae (1,8 Мб [мегабаза]) в 1995 году.[28] В следующем году консорциум исследователей из лабораторий по всему миру. Северная Америка, Европа, и Япония объявил о завершении первой полной последовательности генома эукариота, С. cerevisiae (12,1 Мб), и с тех пор секвенирование геномов продолжается с экспоненциально растущей скоростью.[29] По состоянию на октябрь 2011 г.[Обновить], полные последовательности доступны для: 2 719 вирусы, 1,115 археи и бактерии, и 36 эукариоты, из которых около половины грибы.[30][31]

Большинство микроорганизмов, чьи геномы были полностью секвенированы, проблематичны. патогены, такие как Haemophilus influenzae, что привело к явному смещению их филогенетического распределения по сравнению с широтой микробного разнообразия.[32][33] Из других секвенированных видов большинство было выбрано потому, что они были хорошо изученными модельными организмами или обещали стать хорошими моделями. Дрожжи (Saccharomyces cerevisiae ) долгое время был важным модельный организм для эукариотическая клетка, а плодовая муха Drosophila melanogaster был очень важным инструментом (особенно в раннем домолекулярном генетика ). Червь Caenorhabditis elegans это часто используемая простая модель для многоклеточные организмы. Данио Brachydanio rerio используется для многих исследований развития на молекулярном уровне, и растение Arabidopsis thaliana представляет собой модельный организм для цветущих растений. В Японский иглобрюх (Такифугу рубрипс ) и пятнистый зеленый иглобрюх (Тетраодон нигровиридис ) интересны своими маленькими и компактными геномами, которые содержат очень мало некодирующая ДНК по сравнению с большинством видов.[34][35] Собака млекопитающих (Собаки фамильярные ),[36] коричневая крыса (Раттус норвегикус ), мышь (Mus musculus ) и шимпанзе (Пан троглодиты ) являются важными модельными животными в медицинских исследованиях.[23]
Черновой вариант человеческий геном был завершен Проект генома человека в начале 2001 года, создав много шума.[37] В рамках этого проекта, завершенного в 2003 году, был секвенирован весь геном одного конкретного человека, и к 2007 году эта последовательность была объявлена «завершенной» (менее одной ошибки на 20 000 оснований и собраны все хромосомы).[37] За прошедшие с тех пор годы геномы многих других людей были секвенированы, частично под эгидой Проект 1000 геномов, который объявил о секвенировании 1092 генома в октябре 2012 года.[38] Завершение этого проекта стало возможным благодаря разработке значительно более эффективных технологий секвенирования и потребовало серьезных усилий. биоинформатика ресурсы от большого международного сотрудничества.[39] Постоянный анализ геномных данных человека имеет глубокие политические и социальные последствия для человеческого общества.[40]
Революция "омикс"
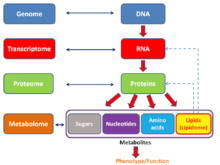
Английский язык неологизм омики неформально относится к области изучения биологии, оканчивающейся на -комикс, например, геномика, протеомика или метаболомика. Связанный суффикс -ome используется для обращения к объектам исследования таких полей, как геном, протеом или метаболом соответственно. Суффикс -ome в молекулярной биологии означает совокупность какой-то; так же омики стал относиться к изучению больших, всеобъемлющих наборов биологических данных. Хотя рост использования этого термина привел к тому, что некоторые ученые (Джонатан Эйзен, среди прочего[41]) заявить, что она была перепродана,[42] он отражает изменение ориентации на количественный анализ полного или почти полного набора всех составляющих системы.[43] При изучении симбиозы Например, исследователи, которые раньше ограничивались изучением одного генного продукта, теперь могут одновременно сравнивать общий набор нескольких типов биологических молекул.[44][45]
Анализ генома
После выбора организма проекты генома включают три компонента: секвенирование ДНК, сборку этой последовательности для создания представления исходной хромосомы, а также аннотацию и анализ этого представления.[6]

Последовательность действий
Исторически секвенирование выполнялось в центры секвенирования, централизованные объекты (начиная от крупных независимых организаций, таких как Объединенный институт генома которые передают десятки терабаз в год на локальные базовые объекты молекулярной биологии), в которых есть исследовательские лаборатории с дорогостоящим оборудованием и необходимой технической поддержкой. Однако по мере совершенствования технологии секвенирования новое поколение эффективных настольных секвенаторов с быстрым оборотом стало доступным для средней академической лаборатории.[46][47] В целом подходы к секвенированию генома делятся на две большие категории: дробовик и высокая пропускная способность (или следующее поколение) последовательность действий.[6]
Секвенирование дробовика

Секвенирование дробовиком - это метод секвенирования, разработанный для анализа последовательностей ДНК длиной более 1000 пар оснований, вплоть до целых хромосом.[48] Он назван по аналогии с быстро расширяющейся квазислучайной схемой срабатывания дробовик. Поскольку секвенирование с помощью гель-электрофореза можно использовать только для довольно коротких последовательностей (от 100 до 1000 пар оснований), более длинные последовательности ДНК должны быть разбиты на случайные небольшие сегменты, которые затем секвенируются для получения читает. Множественные перекрывающиеся считывания для целевой ДНК получают путем выполнения нескольких раундов этой фрагментации и секвенирования. Затем компьютерные программы используют перекрывающиеся концы различных операций чтения, чтобы собрать их в непрерывную последовательность.[48][49] Секвенирование дробовика - это случайный процесс выборки, требующий избыточной выборки для обеспечения заданного нуклеотид представлен в реконструированной последовательности; среднее количество считываний, при которых происходит избыточная выборка генома, называется покрытие.[50]
На протяжении большей части своей истории технология, лежащая в основе секвенирования дробовика, была классическим методом обрыва цепи илиМетод Сенгера ', который основан на избирательном включении обрывающих цепь дидезоксинуклеотиды от ДНК-полимераза в течение in vitro Репликация ДНК.[19][51] В последнее время на смену секвенированию дробовика пришли высокопроизводительное секвенирование методы, особенно для крупномасштабных, автоматизированных геном анализы. Тем не менее, метод Сэнгера по-прежнему широко используется, в первую очередь для небольших проектов и для получения особо длинных считываний непрерывных последовательностей ДНК (> 500 нуклеотидов).[52] Для методов обрыва цепи требуется матрица одноцепочечной ДНК, ДНК грунтовка, а ДНК-полимераза, нормальные дезоксинуклеозидтрифосфаты (дНТФ) и модифицированные нуклеотиды (дидезоксиНТФ), которые останавливают удлинение цепи ДНК. Эти обрывающие цепь нуклеотиды лишены 3'-ОЙ группа, необходимая для формирования фосфодиэфирная связь между двумя нуклеотидами, заставляя ДНК-полимеразу прекращать удлинение ДНК при включении ddNTP. DdNTP могут быть радиоактивными или флуоресцентно помечены для обнаружения в Секвенаторы ДНК.[6] Как правило, эти машины могут секвенировать до 96 образцов ДНК за одну серию (цикл) до 48 циклов в день.[53]
Секвенирование с высокой пропускной способностью
Высокий спрос на дешевое секвенирование стимулировал развитие технологий высокопроизводительного секвенирования, которые распараллеливать процесс секвенирования, производящий одновременно тысячи или миллионы последовательностей.[54][55] Высокопроизводительное секвенирование предназначено для снижения стоимости секвенирования ДНК по сравнению со стандартными методами определения терминатора с использованием красителя. При секвенировании со сверхвысокой пропускной способностью параллельно может выполняться до 500 000 операций секвенирования путем синтеза.[56][57]

В Секвенирование красителей Illumina Метод основан на обратимых терминаторах красителя и был разработан в 1996 году в Женевском институте биомедицинских исследований Паскалем Майером и Лораном Фаринелли.[58] В этом методе молекулы ДНК и праймеры сначала прикрепляются к слайду и амплифицируются с помощью полимераза так что образуются локальные клональные колонии, первоначально названные «колониями ДНК». Для определения последовательности добавляют четыре типа оснований обратимых терминаторов (RT-основания) и смывают невключенные нуклеотиды. В отличие от пиросеквенирования, цепи ДНК удлиняются на один нуклеотид за раз, и получение изображения может выполняться с задержкой, что позволяет захватывать очень большие массивы колоний ДНК с помощью последовательных изображений, полученных с одной камеры. Разделение ферментативной реакции и захвата изображения обеспечивает оптимальную производительность и теоретически неограниченную емкость секвенирования; при оптимальной конфигурации конечная производительность прибора зависит только от Аналого-цифровое преобразование скорость камеры. Камера делает снимки флуоресцентно маркированный нуклеотидов, затем краситель вместе с концевым блокатором 3 'химически удаляется из ДНК, обеспечивая следующий цикл.[59]
Альтернативный подход, ионно-полупроводниковое секвенирование, основан на стандартной химии репликации ДНК. Эта технология измеряет высвобождение иона водорода каждый раз, когда вводится основание. Микролунка, содержащая матричную ДНК, заполняется одним нуклеотид, если нуклеотид комплементарен цепи матрицы, он будет включен и ион водорода будет высвобожден. Этот выпуск вызывает ISFET ионный датчик. Если гомополимер В матричной последовательности присутствует несколько нуклеотидов, которые будут включены в один цикл наводнения, и обнаруженный электрический сигнал будет пропорционально выше.[60]
Сборка
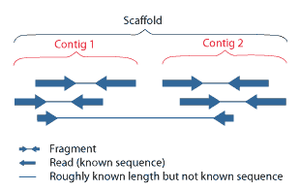
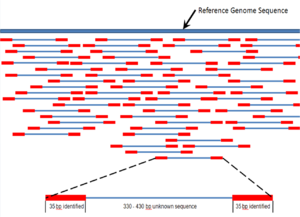
Последовательная сборка относится к выравнивание и слияние фрагментов гораздо более длинного ДНК последовательность, чтобы восстановить исходную последовательность.[6] Это необходимо в качестве текущего Секвенирование ДНК Технология не может считывать целые геномы как непрерывную последовательность, а скорее считывает небольшие фрагменты от 20 до 1000 оснований, в зависимости от используемой технологии. Технологии секвенирования третьего поколения, такие как PacBio или Oxford Nanopore, обычно генерируют чтения секвенирования длиной> 10 т.п.н. однако у них высокий уровень ошибок - примерно 15 процентов.[61][62] Обычно короткие фрагменты, называемые чтениями, являются результатом секвенирование дробовика геномный ДНК, или транскрипты генов (EST ).[6]
Сборочные подходы
Сборку можно условно разделить на два подхода: de novo сборка для геномов, которые не похожи на какие-либо последовательности, секвенированные в прошлом, и сравнительная сборка, при которой в качестве эталона во время сборки используется существующая последовательность тесно связанного организма.[50] Относительно сравнительной сборки, de novo сборка вычислительно трудна (NP-жесткий ), что делает его менее подходящим для короткочитаемых технологий NGS. В рамках de novo В парадигме сборки существуют две основные стратегии сборки: стратегии эйлерова пути и стратегии перекрытия-компоновки-консенсуса (OLC). Стратегии OLC в конечном итоге пытаются создать гамильтонов путь через граф перекрытия, что является NP-трудной задачей. Стратегии эйлерова пути вычислительно более управляемы, потому что они пытаются найти эйлеров путь через граф де Брюйна.[50]
Отделка
Готовые геномы определяются как имеющие одну непрерывную последовательность без неоднозначностей, представляющих каждую репликон.[63]
Аннотации
Сама по себе сборка последовательности ДНК не имеет большого значения без дополнительного анализа.[6] Аннотации генома это процесс присоединения биологической информации к последовательности, и состоит из трех основных шагов:[64]
- идентификация частей генома, которые не кодируют белки
- идентификационные элементы на геном, процесс, называемый предсказание генов, и
- прикрепление биологической информации к этим элементам.
Инструменты автоматического аннотации пытаются выполнить эти шаги in silico, в отличие от ручных аннотаций (также известных как курирование), которые требуют человеческого опыта и потенциальной экспериментальной проверки.[65] В идеале эти подходы сосуществуют и дополняют друг друга в одной аннотации. трубопровод (также см ниже ).
Традиционно базовым уровнем аннотации является использование ВЗРЫВ для поиска сходства, а затем аннотирования геномов на основе гомологов.[6] Совсем недавно на платформу аннотаций добавлена дополнительная информация. Дополнительная информация позволяет ручным аннотаторам деконволютировать несоответствия между генами, которым даны одинаковые аннотации. Некоторые базы данных используют контекстную информацию генома, оценки сходства, экспериментальные данные и интеграцию других ресурсов для предоставления аннотаций генома через свой подход «Подсистемы». Другие базы данных (например, Ансамбль ) полагаются как на тщательно отобранные источники данных, так и на ряд программных инструментов в своем конвейере автоматической аннотации генома.[66] Структурная аннотация состоит из идентификации геномных элементов, в первую очередь ORF и их локализация или структура гена. Функциональная аннотация состоит из присоединения биологической информации к геномным элементам.
Конвейеры секвенирования и базы данных
Необходимость воспроизводимости и эффективного управления большим объемом данных, связанных с проектами генома, означает, что вычислительные конвейеры имеют важные приложения в геномике.[67]
Области исследований
Функциональная геномика
Функциональная геномика это область молекулярная биология который пытается использовать огромное количество данных, полученных в ходе геномных проектов (таких как проекты секвенирования генома ) описать ген (и белок ) функции и взаимодействия. Функциональная геномика фокусируется на динамических аспектах, таких как ген транскрипция, перевод, и белок-белковые взаимодействия, в отличие от статических аспектов геномной информации, таких как Последовательность ДНК или конструкции. Функциональная геномика пытается ответить на вопросы о функции ДНК на уровне генов, транскриптов РНК и белковых продуктов. Ключевой характеристикой исследований функциональной геномики является их общегеномный подход к этим вопросам, как правило, с использованием высокопроизводительных методов, а не более традиционного подхода «ген за геном».
Основная отрасль геномики по-прежнему занимается последовательность действий геномов различных организмов, но знание полных геномов создало возможность для области функциональная геномика, в основном касается моделей экспрессия гена в различных условиях. Самые важные инструменты здесь: микрочипы и биоинформатика.
Структурная геномика
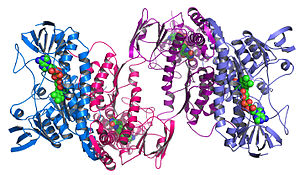
Структурная геномика стремится описать 3-х мерная структура каждого белка, кодируемого данным геном.[68][69] Этот подход, основанный на геноме, позволяет использовать высокопроизводительный метод определения структуры с помощью комбинации экспериментальный и модельный подходы. Принципиальная разница между структурной геномикой и традиционный структурный прогноз состоит в том, что структурная геномика пытается определить структуру каждого белка, кодируемого геномом, а не сосредотачиваться на одном конкретном белке. При наличии полногеномных последовательностей предсказание структуры может быть выполнено быстрее за счет комбинации экспериментального и модельного подходов, особенно потому, что наличие большого количества секвенированных геномов и ранее решенных белковых структур позволяет ученым моделировать структуру белка на структурах ранее решенных. гомологи. Структурная геномика включает использование большого количества подходов к определению структуры, включая экспериментальные методы с использованием геномных последовательностей или подходы на основе моделирования, основанные на последовательности или структурная гомология к белку известной структуры или основанному на химических и физических принципах для белка, не имеющего гомологии с какой-либо известной структурой. В отличие от традиционных структурная биология, определение структура белка через структурную геномику усилия часто (но не всегда) приходят раньше, чем что-либо известно о функции белка. Это ставит новые задачи в структурная биоинформатика, т.е. определение функции белка по его 3D структура.[70]
Эпигеномика
Эпигеномика это исследование полного набора эпигенетический модификации генетического материала клетки, известные как эпигеном.[71] Эпигенетические модификации - это обратимые модификации клеточной ДНК или гистонов, которые влияют на экспрессию генов без изменения последовательности ДНК (Russell 2010, стр. 475). Две из наиболее характерных эпигенетических модификаций: Метилирование ДНК и гистоновая модификация. Эпигенетические модификации играют важную роль в экспрессии и регуляции генов и участвуют во многих клеточных процессах, таких как дифференциация / развитие и туморогенез.[71] Изучение эпигенетики на глобальном уровне стало возможным только недавно благодаря адаптации геномных высокопроизводительных анализов.[72]
Метагеномика
Метагеномика это изучение метагеномы, генетический материал, извлеченный непосредственно из экологический образцы. Это широкое поле может также называться экологической геномикой, экогеномикой или геномикой сообщества. Хотя традиционные микробиология и микробный секвенирование генома полагаться на культивируемые клональный культуры, раннее секвенирование генов окружающей среды клонировало определенные гены (часто 16S рРНК ген), чтобы произвести профиль разнообразия в натуральном образце. Такая работа показала, что подавляющее большинство микробное биоразнообразие был пропущен на основе выращивания методы.[73] Недавние исследования используют «дробовик» Секвенирование по Сэнгеру или массово параллельно пиросеквенирование чтобы получить в значительной степени объективные образцы всех генов от всех членов выбранных сообществ.[74] Благодаря своей способности раскрывать ранее скрытое разнообразие микроскопической жизни, метагеномика предлагает мощную линзу для просмотра микробного мира, которая может революционизировать понимание всего живого мира.[75][76]
Модельные системы
Вирусы и бактериофаги
Бактериофаги сыграли и продолжают играть ключевую роль в бактериальной генетика и молекулярная биология. Исторически они использовались для определения ген структура и генная регуляция. Также первый геном быть упорядоченным был бактериофаг. Однако исследования бактериофагов не привели к революции в геномике, в которой явно доминирует бактериальная геномика.Лишь совсем недавно изучение геномов бактериофагов стало популярным, что позволило исследователям понять механизмы, лежащие в основе фаг эволюция. Последовательности генома бактериофага могут быть получены путем прямого секвенирования изолированных бактериофагов, но также могут быть получены как часть микробных геномов. Анализ бактериальных геномов показал, что значительная часть микробной ДНК состоит из профаг последовательности и профагоподобные элементы.[77] Подробный анализ этих последовательностей в базе данных дает представление о роли профагов в формировании бактериального генома: в целом, этот метод подтвердил многие известные группы бактериофагов, что делает его полезным инструментом для прогнозирования взаимосвязи профагов с геномами бактерий.[78][79]
Цианобактерии
В настоящее время их 24 цианобактерии для которого доступна полная последовательность генома. 15 из этих цианобактерий происходят из морской среды. Это шесть Прохлорококк штаммы, семь морских Синехококк штаммы, Trichodesmium erythraeum IMS101 и Crocosphaera watsonii WH8501. Несколько исследований продемонстрировали, как эти последовательности могут быть очень успешно использованы для вывода важных экологических и физиологических характеристик морских цианобактерий. Однако в настоящее время ведется еще много проектов по геному, среди которых есть и другие. Прохлорококк и морской Синехококк изолирует, Акариохлорис и Прохлор, тогда2-фиксация нитчатых цианобактерий Nodularia spumigena, Lyngbya aestuarii и Lyngbya majuscula, а также бактериофаги заражение морских цианобактерий. Таким образом, растущий объем информации о геноме можно использовать и в более общем плане для решения глобальных проблем, применяя сравнительный подход. Некоторыми новыми и захватывающими примерами прогресса в этой области являются идентификация генов регуляторных РНК, понимание эволюционного происхождения фотосинтез, или оценка вклада горизонтальный перенос генов к геномам, которые были проанализированы.[80]
Приложения геномики
Genomics предлагает приложения во многих областях, в том числе лекарство, биотехнология, антропология и другие социальные науки.[40]
Геномная медицина
Геномные технологии нового поколения позволяют клиницистам и биомедицинским исследователям резко увеличить объем геномных данных, собираемых в больших исследуемых популяциях.[81] В сочетании с новыми подходами к информатике, которые объединяют многие виды данных с геномными данными в исследованиях болезней, это позволяет исследователям лучше понять генетические основы реакции на лекарства и болезни.[82][83] Ранние попытки применить геном в медицине были предприняты командой Стэнфорда во главе с Юан Эшли кто разработал первые инструменты для медицинской интерпретации генома человека.[84][85][86] Например, Все мы Программа исследований направлена на сбор данных о последовательности генома от 1 миллиона участников, чтобы стать важным компонентом исследовательской платформы точной медицины.[87]
Синтетическая биология и биоинженерия
Рост геномных знаний позволил все более изощренные применения синтетическая биология.[88] В 2010 г. исследователи Институт Дж. Крейга Вентера объявил о создании частично синтетического вида бактерия, Лаборатория микоплазм, полученный из геном из Mycoplasma genitalium.[89]
Геномика сохранения
Специалисты по охране природы могут использовать информацию, собранную путем геномного секвенирования, чтобы лучше оценить генетические факторы, ключевые для сохранения видов, такие как генетическое разнообразие популяции или является ли человек гетерозиготным по рецессивному наследственному генетическому заболеванию.[90] Используя геномные данные для оценки эффектов эволюционные процессы и чтобы выявить закономерности в вариациях в данной популяции, защитники природы могут сформулировать планы помощи данному виду, не оставляя неизвестными столько переменных, сколько те, которые не рассматриваются стандартными генетические подходы.[91]
Смотрите также
- Когнитивная геномика
- Вычислительная геномика
- Эпигеномика
- Функциональная геномика
- GeneCalling, технология профилирования мРНК
- Геномика одомашнивания
- Генетика в художественной литературе
- Гликомикс
- Иммуномика
- Метагеномика
- Патогеномика
- Персональная геномика
- Протеомика
- Транскриптомика
- Психогеномика
- Секвенирование всего генома
- Томас Родерик
использованная литература
- ^ «Определения генетики и геномики ВОЗ». Всемирная организация здоровья.
- ^ Национальный институт исследования генома человека (8 ноября 2010 г.). «Краткое руководство по геномике». Genome.gov. Получено 2011-12-03.
- ^ Концепции генетики (10-е изд.). Сан-Франциско: образование Пирсона. 2012 г. ISBN 978-0-321-72412-0.
- ^ Калвер К.В., Лабов М.А. (8 ноября 2002 г.). «Геномика». В Робинзоне R (ред.). Генетика. Научная библиотека Macmillan. Справочник Macmillan USA. ISBN 978-0-02-865606-9.
- ^ Кадаккужа Б.М., Путханветтил С.В. (июль 2013 г.). «Геномика и протеомика в решении сложности мозга». Молекулярные биосистемы. 9 (7): 1807–21. Дои:10.1039 / C3MB25391K. ЧВК 6425491. PMID 23615871.
- ^ а б c d е ж г час я Певснер Дж (2009). Биоинформатика и функциональная геномика (2-е изд.). Хобокен, Нью-Джерси: Уайли-Блэквелл. ISBN 978-0-470-08585-1.
- ^ Лидделл Х.Г., Скотт Р. (1889). Греко-английский лексикон среднего уровня γίγνομαι. Оксфорд: Clarendon Press. ISBN 978-1-61427-397-4. Архивировано из оригинал на 2018-06-20. Получено 2015-05-13.
- ^ "Геном, n". Оксфордский словарь английского языка (Третье изд.). Издательство Оксфордского университета. 2008 г.. Получено 2012-12-01.(требуется подписка)
- ^ Ядав СП (декабрь 2007 г.). "Целостность в суффиксах -omics, -omes и в слове om". Журнал биомолекулярных методов. 18 (5): 277. ЧВК 2392988. PMID 18166670.
- ^ Анкени, РА (июнь 2003 г.). «Секвенирование генома от нематод до человека: изменение методов, изменение науки». Стремление. 27 (2): 87–92. Дои:10.1016 / S0160-9327 (03) 00061-9. PMID 12798815.
- ^ Холли Р.В., Эверетт Г.А., Мэдисон Д.Т., Замир А. (май 1965 г.). «Нуклеотидные последовательности в рибонуклеиновой кислоте, переносящей аланин» (PDF). Журнал биологической химии. 240 (5): 2122–8. PMID 14299636.
- ^ Холли Р.В., Апгар Дж., Эверетт Г.А., Мэдисон Дж. Т., Маркизи М., Меррилл С.Х., Пенсвик Дж. Р., Замир А. (март 1965 г.). «Строение рибонуклеиновой кислоты». Наука. 147 (3664): 1462–5. Bibcode:1965Научный ... 147.1462H. Дои:10.1126 / science.147.3664.1462. PMID 14263761. S2CID 40989800.
- ^ Ниренберг М., Ледер П., Бернфилд М., Бримакомб Р., Трупин Дж., Роттман Ф., О'Нил С. (май 1965 г.). «Кодовые слова РНК и синтез белка, VII. Об общей природе кода РНК». Труды Национальной академии наук Соединенных Штатов Америки. 53 (5): 1161–8. Bibcode:1965ПНАС ... 53.1161Н. Дои:10.1073 / pnas.53.5.1161. ЧВК 301388. PMID 5330357.
- ^ Мин Джоу В., Хэгеман Г., Исебаерт М., Фирс В. (май 1972 г.). «Нуклеотидная последовательность гена, кодирующего белок оболочки бактериофага MS2». Природа. 237 (5350): 82–8. Bibcode:1972 год. 237 ... 82J. Дои:10.1038 / 237082a0. PMID 4555447. S2CID 4153893.
- ^ Fiers W, Contreras R, Duerinck F, Haegeman G, Iserentant D, Merregaert J, et al. (Апрель 1976 г.). «Полная нуклеотидная последовательность РНК бактериофага MS2: первичная и вторичная структура гена репликазы». Природа. 260 (5551): 500–7. Bibcode:1976Натура.260..500F. Дои:10.1038 / 260500a0. PMID 1264203. S2CID 4289674.
- ^ Фирс В., Контрерас Р., Хегеманн Г., Роджерс Р., Ван де Вурде А., Ван Хеуверсвин Х., Ван Херревеге Дж., Фолькаерт Г., Исебаерт М. (май 1978 г.). «Полная нуклеотидная последовательность ДНК SV40». Природа. 273 (5658): 113–20. Bibcode:1978Натура.273..113F. Дои:10.1038 / 273113a0. PMID 205802. S2CID 1634424.
- ^ Тамарин Р.Х. (2004). Принципы генетики (7-е изд.). Лондон: Макгроу Хилл. ISBN 978-0-07-124320-9.
- ^ Сэнгер Ф (1980). «Нобелевская лекция: Определение нуклеотидных последовательностей в ДНК» (PDF). Nobelprize.org. Получено 2010-10-18.
- ^ а б Сэнгер Ф., Air GM, Баррелл Б.Г., Браун Н.Л., Коулсон А.Р., Фиддес, Калифорния, Хатчисон, Калифорния, Слокомб П.М., Смит М. (февраль 1977 г.). «Нуклеотидная последовательность ДНК бактериофага phi X174». Природа. 265 (5596): 687–95. Bibcode:1977Натура.265..687С. Дои:10.1038 / 265687a0. PMID 870828. S2CID 4206886.
- ^ Kaiser O, Bartels D, Bekel T, Goesmann A, Kespohl S, Pühler A, Meyer F (декабрь 2003 г.). «Полное геномное секвенирование с помощью биоинформатических конвейеров - оптимизированный подход для установленной техники». Журнал биотехнологии. 106 (2–3): 121–33. Дои:10.1016 / j.jbiotec.2003.08.008. PMID 14651855.
- ^ Сэнгер Ф., Никлен С, Колсон А.Р. (декабрь 1977 г.). «Секвенирование ДНК с помощью ингибиторов обрыва цепи». Труды Национальной академии наук Соединенных Штатов Америки. 74 (12): 5463–7. Bibcode:1977ПНАС ... 74.5463С. Дои:10.1073 / pnas.74.12.5463. ЧВК 431765. PMID 271968.
- ^ Максам AM, Гилберт В. (февраль 1977 г.). «Новый метод секвенирования ДНК». Труды Национальной академии наук Соединенных Штатов Америки. 74 (2): 560–4. Bibcode:1977ПНАС ... 74..560М. Дои:10.1073 / пнас.74.2.560. ЧВК 392330. PMID 265521.
- ^ а б Дарден Л., Джеймс Табери (2010). "Молекулярная биология". В Залте EN (ред.). Стэнфордская энциклопедия философии (Издание осенью 2010 г.).
- ^ Андерсон С., Банкир А.Т., Баррелл Б.Г., де Брейн М.Х., Колсон А.Р., Друин Дж. И др. (Апрель 1981 г.). «Последовательность и организация митохондриального генома человека». Природа. 290 (5806): 457–65. Bibcode:1981Натура.290..457A. Дои:10.1038 / 290457a0. PMID 7219534. S2CID 4355527.(требуется подписка)
- ^ Шинозаки К., Оме М., Танака М., Вакасуги Т., Хаяшида Н., Мацубаяси Т. и др. (Сентябрь 1986 г.). «Полная нуклеотидная последовательность генома хлоропласта табака: его генная организация и экспрессия». Журнал EMBO. 5 (9): 2043–2049. Дои:10.1002 / j.1460-2075.1986.tb04464.x. ЧВК 1167080. PMID 16453699.
- ^ Охьяма К., Фукудзава Х., Кохчи Т., Шираи Х., Сано Т., Сано С. и др. (1986). "Организация гена хлоропласта выведена из полной последовательности ДНК хлоропласта печеночника Marchantia polymorpha". Природа. 322 (6079): 572–574. Bibcode:1986Натура.322..572O. Дои:10.1038 / 322572a0. S2CID 4311952.
- ^ Оливер С.Г., van der Aart QJ, Agostoni-Carbone ML, Aigle M, Alberghina L, Alexandraki D, Antoine G, Anwar R, Ballesta JP, Benit P (май 1992 г.). «Полная последовательность ДНК дрожжевой хромосомы III». Природа. 357 (6373): 38–46. Bibcode:1992Натура 357 ... 38О. Дои:10.1038 / 357038a0. PMID 1574125. S2CID 4271784.
- ^ Fleischmann RD, Adams MD, White O, Clayton RA, Kirkness EF, Kerlavage AR и др. (Июль 1995 г.). «Полногеномное случайное секвенирование и сборка Haemophilus influenzae Rd». Наука. 269 (5223): 496–512. Bibcode:1995Научный ... 269..496F. Дои:10.1126 / science.7542800. PMID 7542800. S2CID 10423613.
- ^ Гоффо А., Баррелл Б.Г., Бусси Х., Дэвис Р.В., Дуйон Б., Фельдманн Х., Галиберт Ф., Хохейзель Д.Д., Жак С., Джонстон М., Луис Э.Дж., Мьюз Х.В., Мураками И., Филиппсен П., Теттелин Х., Оливер С.Г. (октябрь 1996 г. ). «Жизнь с 6000 генами». Наука. 274 (5287): 546, 563–7. Bibcode:1996Наука ... 274..546G. Дои:10.1126 / science.274.5287.546. PMID 8849441. S2CID 211123134.(требуется подписка)
- ^ «Полные геномы: вирусы». NCBI. 17 ноября 2011 г.. Получено 2011-11-18.
- ^ «Статистика геномного проекта». Энтрез Геном Проект. 7 октября 2011 г.. Получено 2011-11-18.
- ^ Циммер С. (29 декабря 2009 г.). «Ученые создают геномный каталог многочисленных микробов Земли». Нью-Йорк Таймс. ISSN 0362-4331. Получено 2012-12-21.
- ^ Ву Д., Гугенгольц П., Мавроматис К., Пукалл Р., Далин Е., Иванова Н. Н. и др. (Декабрь 2009 г.). «Филогенетическая геномная энциклопедия бактерий и архей». Природа. 462 (7276): 1056–60. Bibcode:2009 Натур.462.1056W. Дои:10.1038 / природа08656. ЧВК 3073058. PMID 20033048.
- ^ "Число генов человека сокращено". BBC. 20 октября 2004 г.. Получено 2012-12-21.
- ^ Юэ GH, Lo LC, Zhu ZY, Lin G, Feng F (апрель 2006 г.). «Полная нуклеотидная последовательность митохондриального генома Tetraodon nigroviridis». Последовательность ДНК. 17 (2): 115–21. Дои:10.1080/10425170600700378. PMID 17076253. S2CID 21797344.
- ^ Национальный институт исследования генома человека (14 июля 2004 г.). «Геном собаки собран: геном собаки теперь доступен для исследовательского сообщества во всем мире». Genome.gov. Получено 2012-01-20.
- ^ а б Макэлхени V (2010). Рисуем карту жизни: внутри проекта "Геном человека". Нью-Йорк Нью-Йорк: Основные книги. ISBN 978-0-465-04333-0.
- ^ Абекасис Г. Р., Аутон А., Брукс Л. Д., ДеПристо М. А., Дурбин Р. М., Хандакер Р. Э., Канг Х. М., Март Г. Т., Маквин Г. А. (ноябрь 2012 г.). «Интегрированная карта генетических вариаций из 1092 геномов человека». Природа. 491 (7422): 56–65. Bibcode:2012Натура 491 ... 56Т. Дои:10.1038 / природа11632. ЧВК 3498066. PMID 23128226.
- ^ Нильсен Р. (октябрь 2010 г.). «Геномика: в поисках редких вариантов человека». Природа. 467 (7319): 1050–1. Bibcode:2010Натура.467.1050N. Дои:10.1038 / 4671050a. PMID 20981085.
- ^ а б Барнс Б., Дюпре Дж. (2008). Геномы и что с ними делать. Чикаго: Издательство Чикагского университета. ISBN 978-0-226-17295-8.
- ^ Эйзен Дж. А. (июль 2012 г.). «Бадомические слова, сила и опасность омэ-мема». GigaScience. 1 (1): 6. Дои:10.1186 / 2047-217X-1-6. ЧВК 3617454. PMID 23587201.
- ^ Хотц Р.Л. (13 августа 2012 г.). "Вот химическая сказка: ученые открывают распространяющийся суффикс". Wall Street Journal. ISSN 0099-9660. Получено 2013-01-04.
- ^ Scudellari M (1 октября 2011 г.). "Поток данных". Ученый. Получено 2013-01-04.
- ^ Честон Дж., Дуглас А.Е. (август 2012 г.). "Максимальное использование" омиков "для исследования симбиоза". Биологический бюллетень. 223 (1): 21–9. Дои:10.1086 / BBLv223n1p21. ЧВК 3491573. PMID 22983030.
- ^ McCutcheon JP, von Dohlen CD (август 2011 г.). «Взаимозависимый метаболический пэчворк во вложенном симбиозе мучнистых червецов». Текущая биология. 21 (16): 1366–72. Дои:10.1016 / j.cub.2011.06.051. ЧВК 3169327. PMID 21835622.
- ^ а б Бейкер М (14 сентября 2012 г.). "Настольные секвенсоры отгружаются" (Блог). Блог новостей природы. Получено 2012-12-22.
- ^ Перепел М.А., Смит М., Коупленд П., Отто Т.Д., Харрис С.Р., Коннор Т.Р., Бертони А., Свердлоу Х.П., Гу Y (июль 2012 г.). «Рассказ о трех платформах секвенирования следующего поколения: сравнение секвенсоров Ion Torrent, Pacific Biosciences и Illumina MiSeq». BMC Genomics. 13: 341. Дои:10.1186/1471-2164-13-341. ЧВК 3431227. PMID 22827831.
- ^ а б Staden R (июнь 1979 г.). «Стратегия секвенирования ДНК с использованием компьютерных программ». Исследования нуклеиновых кислот. 6 (7): 2601–10. Дои:10.1093 / nar / 6.7.2601. ЧВК 327874. PMID 461197.
- ^ Андерсон С. (июль 1981 г.). «Секвенирование ДНК дробовика с использованием клонированных фрагментов ДНКазы I». Исследования нуклеиновых кислот. 9 (13): 3015–27. Дои:10.1093 / nar / 9.13.3015. ЧВК 327328. PMID 6269069.
- ^ а б c Pop M (июль 2009 г.). «Возрождение сборки генома: недавние вычислительные проблемы». Брифинги по биоинформатике. 10 (4): 354–66. Дои:10.1093 / bib / bbp026. ЧВК 2691937. PMID 19482960.
- ^ Сэнгер Ф., Колсон А.Р. (май 1975 г.). «Экспресс-метод определения последовательностей в ДНК путем примированного синтеза с ДНК-полимеразой». Журнал молекулярной биологии. 94 (3): 441–8. Дои:10.1016/0022-2836(75)90213-2. PMID 1100841.
- ^ Мавроматис К., Лэнд М.Л., Бреттин Т.С., Quest DJ, Коупленд А., Клам А. и др. (2012). Лю Z (ред.). «Быстро меняющийся ландшафт технологий секвенирования и их влияние на сборки и аннотации микробного генома». PLOS ONE. 7 (12): e48837. Bibcode:2012PLoSO ... 748837M. Дои:10.1371 / journal.pone.0048837. ЧВК 3520994. PMID 23251337.
- ^ Illumina, Inc. (28 февраля 2012 г.). Введение в технологию секвенирования нового поколения (PDF). Сан-Диего, Калифорния, США: Illumina, Inc. стр. 12. Получено 2012-12-28.
- ^ Зал N (май 2007 г.). «Передовые технологии секвенирования и их более широкое влияние на микробиологию». Журнал экспериментальной биологии. 210 (Pt 9): 1518–25. Дои:10.1242 / jeb.001370. PMID 17449817.
- ^ Церковь GM (январь 2006 г.). «Геномы для всех». Scientific American. 294 (1): 46–54. Bibcode:2006SciAm.294a..46C. Дои:10.1038 / scientificamerican0106-46. PMID 16468433.
- ^ ten Bosch JR, Grody WW (ноябрь 2008 г.). «Идти в ногу со следующим поколением: массовое параллельное секвенирование в клинической диагностике». Журнал молекулярной диагностики. 10 (6): 484–92. Дои:10.2353 / jmoldx.2008.080027. ЧВК 2570630. PMID 18832462.
- ^ Такер Т., Марра М., Фридман Дж. М. (август 2009 г.). «Массовое параллельное секвенирование: следующий большой шаг в генетической медицине». Американский журнал генетики человека. 85 (2): 142–54. Дои:10.1016 / j.ajhg.2009.06.022. ЧВК 2725244. PMID 19679224.
- ^ Кавасима Э., Фаринелли Л., Майер П. (12 мая 2005 г.). «Метод амплификации нуклеиновых кислот». Получено 2012-12-22.
- ^ Мардис ER (2008). «Методы секвенирования ДНК нового поколения» (PDF). Ежегодный обзор геномики и генетики человека. 9: 387–402. Дои:10.1146 / annurev.genom.9.081307.164359. PMID 18576944. Архивировано из оригинал (PDF) на 2013-05-18. Получено 2013-01-04.
- ^ Дэвис К. (2011). «Эффективная профилактическая медицина». Биотехнологический мир (Сентябрь октябрь).
- ^ https://www.pacb.com/
- ^ "Оксфорд Нанопор Технолоджис".
- ^ Chain PS, Grafham DV, Fulton RS, Fitzgerald MG, Hostetler J, Muzny D, et al. (Октябрь 2009 г.). «Геномика. Стандарты геномных проектов в новую эру секвенирования». Наука. 326 (5950): 236–7. Bibcode:2009Sci ... 326..236C. Дои:10.1126 / science.1180614. ЧВК 3854948. PMID 19815760.
- ^ Штейн Л. (июль 2001 г.). «Аннотации генома: от последовательности к биологии». Обзоры природы. Генетика. 2 (7): 493–503. Дои:10.1038/35080529. PMID 11433356. S2CID 12044602.
- ^ Brent MR (январь 2008 г.). «Устойчивый прогресс и недавние прорывы в точности автоматизированной аннотации генома» (PDF). Обзоры природы. Генетика. 9 (1): 62–73. Дои:10.1038 / nrg2220. PMID 18087260. S2CID 20412451. Архивировано из оригинал (PDF) на 2013-05-29. Получено 2013-01-04.
- ^ Фличек П., Ахмед И., Амод М.Р., Баррелл Д., Бил К., Брент С. и др. (Январь 2013). «Ансамбль 2013». Исследования нуклеиновых кислот. 41 (Выпуск базы данных): D48–55. Дои:10.1093 / нар / гкс1236. ЧВК 3531136. PMID 23203987.
- ^ Кейт Дж. М. (2008). Кейт Дж. М. (ред.). Биоинформатика. Методы молекулярной биологии. 453. стр. v – vi. Дои:10.1007/978-1-60327-429-6. ISBN 978-1-60327-428-9. PMID 18720577.
- ^ Марсден Р.Л., Льюис Т.А., Оренго, Калифорния (март 2007 г.). «На пути к полному структурному охвату завершенных геномов: взгляд на структурную геномику». BMC Bioinformatics. 8: 86. Дои:10.1186/1471-2105-8-86. ЧВК 1829165. PMID 17349043.
- ^ Бреннер С.Е., Левитт М. (январь 2000 г.). «Ожидания от структурной геномики». Белковая наука. 9 (1): 197–200. Дои:10.1110 / пс.9.1.197. ЧВК 2144435. PMID 10739263.
- ^ Бреннер С.Е. (октябрь 2001 г.). «Тур по структурной геномике» (PDF). Обзоры природы. Генетика. 2 (10): 801–9. Дои:10.1038/35093574. PMID 11584296. S2CID 5656447.
- ^ а б Фрэнсис RC (2011). Эпигенетика: высшая загадка наследования. Нью-Йорк: У.В. Нортон. ISBN 978-0-393-07005-7.
- ^ Laird PW (март 2010 г.). «Принципы и проблемы анализа метилирования ДНК в геноме». Обзоры природы. Генетика. 11 (3): 191–203. Дои:10.1038 / nrg2732. PMID 20125086. S2CID 6780101.
- ^ Hugenholtz P, Goebel BM, Pace NR (сентябрь 1998 г.). «Влияние культурно-независимых исследований на формирующееся филогенетическое представление о бактериальном разнообразии». Журнал бактериологии. 180 (18): 4765–74. Дои:10.1128 / JB.180.18.4765-4774.1998. ЧВК 107498. PMID 9733676.
- ^ Эйзен Дж. А. (март 2007 г.). «Экологическое секвенирование ружья: его потенциал и проблемы для изучения скрытого мира микробов». PLOS Биология. 5 (3): e82. Дои:10.1371 / journal.pbio.0050082. ЧВК 1821061. PMID 17355177.
- ^ Марко Д., изд. (2010). Метагеномика: теория, методы и приложения. Caister Academic Press. ISBN 978-1-904455-54-7.
- ^ Марко Д., изд. (2011). Метагеномика: современные инновации и будущие тенденции. Caister Academic Press. ISBN 978-1-904455-87-5.
- ^ Canchaya C, Proux C, Fournous G, Bruttin A, Brüssow H (июнь 2003 г.). «Профагеномика». Обзоры микробиологии и молекулярной биологии. 67 (2): 238–76, содержание. Дои:10.1128 / MMBR.67.2.238-276.2003. ЧВК 156470. PMID 12794192.
- ^ McGrath S, van Sinderen D, ред. (2007). Бактериофаг: генетика и молекулярная биология (1-е изд.). Caister Academic Press. ISBN 978-1-904455-14-1.
- ^ Fouts DE (ноябрь 2006 г.). «Phage_Finder: автоматическая идентификация и классификация областей профага в полных последовательностях бактериального генома». Исследования нуклеиновых кислот. 34 (20): 5839–51. Дои:10.1093 / нар / gkl732. ЧВК 1635311. PMID 17062630.
- ^ Эрреро А., Флорес Э., ред. (2008). Цианобактерии: молекулярная биология, геномика и эволюция (1-е изд.). Caister Academic Press. ISBN 978-1-904455-15-8.
- ^ Hudson KL (сентябрь 2011 г.). «Геномика, здравоохранение и общество». Медицинский журнал Новой Англии. 365 (11): 1033–41. Дои:10.1056 / NEJMra1010517. PMID 21916641.
- ^ О'Доннелл CJ, Набель EG (декабрь 2011 г.). «Геномика сердечно-сосудистых заболеваний». Медицинский журнал Новой Англии. 365 (22): 2098–109. Дои:10.1056 / NEJMra1105239. PMID 22129254.
- ^ Лу Ю.Ф., Гольдштейн ДБ, Ангрист М., Каваллери Г. (июль 2014 г.). «Персонализированная медицина и генетическое разнообразие человека». Перспективы Колд-Спринг-Харбор в медицине. 4 (9): a008581. Дои:10.1101 / cshperspect.a008581. ЧВК 4143101. PMID 25059740.
- ^ Эшли, Юан А; Бьютт, Атул Дж; Уиллер, Мэтью Т; Чен, Ронг; Klein, Teri E; Дьюи, Фредерик Э; Дадли, Джоэл Т; Ормонд, Келли Э; Павлович, Александра; Морган, Александр А; Пушкарев Дмитрий; Нефф, Норма Ф; Хаджинс, Луанн; Гонг, Ли; Ходжес, Лаура М; Берлин, Дорит С; Торн, Кэролайн Ф; Сангкуль, Катрин; Hebert, Joan M; Вун, Марк; Сагрейя, Херш; Уэйли, Райан; Ноулз, Джошуа В.; Чоу, Майкл Ф; Такурия, Джозеф V; Розенбаум, Авраам М; Заранек, Александр Ждут; Церковь, Джордж М; Грили, Генри Т; Quake, Стивен Р.; Альтман, Расс Б. (май 2010 г.). «Клиническая оценка с использованием личного генома». Ланцет. 375 (9725): 1525–1535. Дои:10.1016 / S0140-6736 (10) 60452-7. ЧВК 2937184. PMID 20435227.
- ^ Дьюи, Фредерик Э .; Чен, Ронг; Cordero, Sergio P .; Ормонд, Келли Э .; Калешу, Коллин; Karczewski, Konrad J .; Whirl-Carrillo, Мишель; Уиллер, Мэтью Т .; Дадли, Джоэл Т .; Бирнс, Джейк К .; Cornejo, Omar E .; Ноулз, Джошуа В .; Вун, Марк; Сангкуль, Катрин; Гонг, Ли; Торн, Кэролайн Ф .; Hebert, Joan M .; Каприотти, Эмидио; Дэвид, Шон П .; Павлович, Александра; Уэст, Энн; Такурия, Джозеф V .; Болл, Мадлен П .; Заранек, Александр В .; Rehm, Heidi L .; Церковь, Джордж М .; West, John S .; Bustamante, Carlos D .; Снайдер, Майкл; Альтман, Русь Б .; Klein, Teri E .; Butte, Atul J .; Эшли, Юан А. (15 сентября 2011 г.). «Поэтапный геномный риск для всего генома в семейном квартете с использованием эталонной последовательности мажорного аллеля». PLOS Genetics. 7 (9): e1002280. Дои:10.1371 / journal.pgen.1002280. ЧВК 3174201. PMID 21935354.
- ^ Дьюи, Фредерик Э .; Grove, Megan E .; Пан, Cuiping; Гольдштейн, Бенджамин А .; Бернштейн, Джонатан А .; Хаиб, Хасан; Меркер, Джейсон Д .; Голдфедер, Рэйчел Л .; Эннс, Грегори М .; Дэвид, Шон П .; Пакдаман, Неда; Ормонд, Келли Э .; Калешу, Коллин; Кингхэм, Керри; Klein, Teri E .; Whirl-Carrillo, Мишель; Сакамото, Кеннет; Уиллер, Мэтью Т .; Butte, Atul J .; Форд, Джеймс М .; Боксёр Линда; Иоаннидис, Джон П. А .; Юнг, Алан С .; Альтман, Русь Б .; Assimes, Themistocles L .; Снайдер, Майкл; Ashley, Euan A .; Квертермус, Томас (12 марта 2014 г.). «Клиническая интерпретация и значение секвенирования всего генома». JAMA. 311 (10): 1035–45. Дои:10.1001 / jama.2014.1717. ЧВК 4119063. PMID 24618965.
- ^ "Центры генома, финансируемые Национальным институтом здравоохранения, для ускорения открытий в области точной медицины". Национальные институты здравоохранения: программа исследований для всех нас. Национальные институты здоровья.
- ^ Церковь GM, Regis E (2012). Регенезис: как синтетическая биология изменит природу и нас самих. Нью-Йорк: Основные книги. ISBN 978-0-465-02175-8.
- ^ Бейкер М (май 2011 г.). «Синтетические геномы: следующий шаг на пути к синтетическому геному». Природа. 473 (7347): 403, 405–8. Bibcode:2011Натура.473..403Б. Дои:10.1038 / 473403a. PMID 21593873. S2CID 205064528.
- ^ Фрэнкэм Р. (1 сентября 2010 г.). «Вызовы и возможности генетических подходов к биологической консервации». Биологическое сохранение. 143 (9): 1922–1923. Дои:10.1016 / j.biocon.2010.05.011.
- ^ Аллендорф Ф.В., Гогенлоэ П.А., Луйкарт Г. (октябрь 2010 г.). «Геномика и будущее генетики сохранения». Обзоры природы. Генетика. 11 (10): 697–709. Дои:10.1038 / nrg2844. PMID 20847747. S2CID 10811958.
дальнейшее чтение
- Леск А.М. (2017). Введение в геномику (3-е изд.). Нью-Йорк: Издательство Оксфордского университета. п. 544. ISBN 978-0-19-107085-3. КАК В 0198754833.
- Штунненберг Х.Г., Хубнер Северная Каролина (2014). «Геномика встречается с протеомикой: определение виновников болезни». Генетика человека. 133 (6): 689–700. Дои:10.1007 / s00439-013-1376-2. ЧВК 4021166. PMID 24135908.
- Шибата Т. (2012). «Геномика рака и патология: теперь все вместе». Патология Интернэшнл. 62 (10): 647–59. Дои:10.1111 / j.1440-1827.2012.02855.x. PMID 23005591. S2CID 27886018.
- Ройчоудхури С., Чиннайян А.М. (2016). «Перевод геномов и транскриптомов рака для точной онкологии». КА: Журнал онкологических заболеваний для клиницистов. 66 (1): 75–88. Дои:10.3322 / caac.21329. ЧВК 4713245. PMID 26528881.
- Вадим Н.Г., Чжан Ю. (2013). "Глава 16 Сравнительный геномный анализ металломов". В Banci L (ред.). Металломика и клетка. Ионы металлов в науках о жизни. 12. Springer. Дои:10.1007/978-94-007-5561-10_16 (неактивно 09.11.2020). ISBN 978-94-007-5560-4.CS1 maint: DOI неактивен по состоянию на ноябрь 2020 г. (ссылка на сайт) электронная книга ISBN 978-94-007-5561-1 ISSN 1559-0836 электронный-ISSN 1868-0402
внешние ссылки
- Ежегодный обзор геномики и генетики человека
- BMC Genomics: Журнал BMC по геномике
- Журнал геномики
- Genomics.org: Бесплатный портал по геномике.
- NHGRI: Институт генома правительства США
- Комплексный микробный ресурс JCVI
- KoreaGenome.org: Опубликован первый корейский геном, и последовательность доступна бесплатно.
- GenomicsNetwork: Рассматривает развитие и использование науки и технологий геномики.
- Институт геномных наук: Геномические исследования.
- MIT OpenCourseWare HST.512 Геномная медицина Бесплатный курс самообучения по геномной медицине. Ресурсы включают аудиолекции и избранные конспекты лекций.
- ENCODE Thread Explorer Подходы машинного обучения к геномике. Природа (журнал)
- Глобальная карта лабораторий геномики
- Геномика: научное образование от природы
| Геномика | |
|---|---|
| Биоинформатика | |
| Структурная биология | |
| Инструменты исследования | |
| Организации |
|
| |
| Ключевые компоненты | |
|---|---|
| Поля | |
| Археогенетика из | |
| похожие темы | |
| Списки | |
| |
| люди | |
|---|---|
| Общества | |
| Проекты | |
| Сервисы | |
| |