Метагеномика - Википедия - Metagenomics

Метагеномика это изучение генетический материал, извлеченный непосредственно из относящийся к окружающей среде образцы. Широкое поле можно также обозначить как экологическая геномика, экогеномика или же геномика сообщества.
Хотя традиционные микробиология и микробный секвенирование генома и геномика полагаться на культивируемые клональный культуры, раннее секвенирование генов окружающей среды клонировало определенные гены (часто 16S рРНК ген) для получения профиля разнообразия в естественном образце. Такая работа показала, что подавляющее большинство микробное биоразнообразие были упущены методами выращивания.[2]
Благодаря своей способности раскрывать ранее скрытое разнообразие микроскопической жизни, метагеномика предлагает мощную линзу для наблюдения за микробным миром, которая может революционизировать понимание всего живого мира.[3] Поскольку цена секвенирования ДНК продолжает падать, метагеномика теперь позволяет микробная экология должны быть исследованы в гораздо большем масштабе и детализации, чем раньше. Недавние исследования используют либо "дробовик " или же ПЦР направленное секвенирование для получения в значительной степени объективных выборок всех генов от всех членов выбранных сообществ.[4]
Этимология
Термин «метагеномика» впервые был использован Джо Хандельсман, Джон Кларди, Роберт М. Гудман, Шон Ф. Брэди и другие, и впервые появилось в публикации в 1998 году.[5] Термин «метагеном» относится к идее о том, что набор генов, секвенированных из окружающей среды, может быть проанализирован аналогично исследованию одного геном. В 2005 году Кевин Чен и Лиор Пахтер (исследователи из Калифорнийский университет в Беркли ) определил метагеномику как «применение современной техники геномики без необходимости изолирования и лабораторного культивирования отдельных видов».[6]
История
Общепринятый последовательность действий начинается с культивирования идентичных клеток в качестве источника ДНК. Однако ранние метагеномные исследования показали, что во многих средах, вероятно, существуют большие группы микроорганизмов, которые нельзя культурный и поэтому не может быть упорядочен. Эти ранние исследования были сосредоточены на 16S. рибосомальный РНК (рРНК) относительно короткие, часто консервированный внутри вида и, как правило, различаются между видами. Многие 16S рРНК были обнаружены последовательности, не принадлежащие ни к одному из известных культивируемых разновидность, что указывает на то, что существует множество неизолированных организмов. Эти исследования генов рибосомной РНК, взятых непосредственно из окружающей среды, показали, что выращивание методы, основанные на обнаружении менее 1% бактериальных и архей виды в выборке.[2] Большой интерес к метагеномике проистекает из этих открытий, которые показали, что подавляющее большинство микроорганизмов ранее оставалось незамеченным.
Рано молекулярная работа в поле был проведен Норман Р. Пейс и коллеги, которые использовали ПЦР изучить разнообразие последовательностей рибосомных РНК.[7] Понимание, полученное в результате этих революционных исследований, привело Пейса к предложению идеи клонирования ДНК непосредственно из образцов окружающей среды еще в 1985 году.[8] Это привело к первому сообщению об изоляции и клонирование объемная ДНК из образца окружающей среды, опубликованная Пейсом и его коллегами в 1991 г.[9] в то время как Пейс был на кафедре биологии в Университет Индианы. Значительные усилия гарантировали, что это не ПЦР ложные срабатывания и подтверждали существование сложного сообщества неизведанных видов. Хотя эта методология ограничивалась изучением высококонсервных, гены, не кодирующие белки, это подтвердило ранние наблюдения, основанные на морфологии микробов, о том, что разнообразие было гораздо более сложным, чем было известно методами культивирования. Вскоре после этого Хили сообщил о метагеномном выделении функциональных генов из «зообиблиотек», созданных из сложной культуры организмов окружающей среды, выращенных в лаборатории на сушеных травы в 1995 г.[10] Покинув лабораторию Пейс, Эдвард Делонг продолжил в этой области и опубликовал работу, которая в значительной степени заложила основу экологической филогении на основе сигнатурных последовательностей 16S, начиная с создания его группой библиотек из морской образцы.[11]
В 2002, Mya Breitbart, Forest Rohwer, и его коллеги использовали секвенирование окружающей среды (см. ниже), чтобы показать, что 200 литров морской воды содержат более 5000 различных вирусов.[12] Последующие исследования показали, что существует более тысячи вирусные виды в стуле человека и, возможно, миллион различных вирусов на килограмм морских отложений, в том числе многие бактериофаги. По сути, все вирусы в этих исследованиях были новыми видами. В 2004 году Джин Тайсон, Джилл Бэнфилд и его коллеги из Калифорнийский университет в Беркли и Объединенный институт генома секвенированная ДНК, извлеченная из кислотный дренаж шахты система.[13] Эти усилия привели к созданию полных или почти полных геномов горстки бактерий и археи которые ранее сопротивлялись попыткам их культивирования.[14]
Начиная с 2003 г., Крейг Вентер, лидер частной параллели Проект "Геном человека", возглавил Глобальная экспедиция по отбору проб океана (GOS), совершая кругосветное путешествие и собирая метагеномные образцы на протяжении всего путешествия. Все эти образцы секвенируются с использованием дробовика в надежде, что будут идентифицированы новые геномы (и, следовательно, новые организмы). Пилотный проект, проведенный в г. Саргассово море, нашли ДНК почти 2000 различных разновидность, в том числе 148 видов бактерии никогда раньше не видел.[15] Вентер совершил кругосветное путешествие и тщательно изучил Западное побережье США, и завершил двухлетнюю экспедицию по исследованию Балтийский, Средиземноморье и Чернить Моря. Анализ метагеномных данных, собранных во время этого путешествия, выявил две группы организмов, одна из которых состоит из таксонов, адаптированных к условиям окружающей среды `` пир или голод '', а вторая состоит из относительно меньшего, но более многочисленного и широко распространенного таксона, в основном состоящего из планктон.[16]
В 2005 году Стефан Шустер в Государственный университет Пенсильвании и его коллеги опубликовали первые последовательности образца окружающей среды, созданного с помощью высокопроизводительное секвенирование, в этом случае массивно параллельные пиросеквенирование разработан 454 Науки о жизни.[17] Еще одна ранняя статья в этой области появилась в 2006 году Робертом Эдвардсом, Forest Rohwer и коллеги из Государственный университет Сан-Диего.[18]
Последовательность действий

Восстановление последовательностей ДНК длиной более нескольких тысяч пар оснований из окружающей среды образцы было очень сложно до недавних достижений в молекулярно-биологический методы позволили построить библиотеки в бактериальные искусственные хромосомы (BACs), которые обеспечивали лучшее векторов за молекулярное клонирование.[20]
Метагеномика дробовика
Достижения в биоинформатика, усовершенствования амплификации ДНК и увеличение вычислительной мощности в значительной степени помогли анализу последовательностей ДНК, извлеченных из образцов окружающей среды, что позволило адаптировать секвенирование дробовика к метагеномным образцам (известный также как дробовик целого метагенома или секвенирование WMGS). Подход, используемый для секвенирования многих культивируемых микроорганизмов и человеческий геном, случайным образом срезает ДНК, выстраивает множество коротких последовательностей и реконструирует их в консенсусная последовательность. Секвенирование с дробовиком выявляет гены, присутствующие в образцах окружающей среды. Исторически для облегчения этого секвенирования использовались библиотеки клонов. Однако с развитием технологий высокопроизводительного секвенирования этап клонирования больше не требуется, и можно получить больший объем данных секвенирования без этого трудоемкого этапа узких мест. Метагеномика дробовика предоставляет информацию как о том, какие организмы присутствуют, так и о том, какие метаболические процессы возможны в сообществе.[21] Поскольку сбор ДНК из окружающей среды в значительной степени неконтролируемый, наиболее распространенные организмы в образце окружающей среды наиболее полно представлены в результирующих данных последовательности. Для достижения высокого охвата, необходимого для полного определения геномов недостаточно представленных членов сообщества, необходимы большие выборки, часто даже непомерно высокие. С другой стороны, случайный характер секвенирования с дробовиком гарантирует, что многие из этих организмов, которые в противном случае остались бы незамеченными при использовании традиционных методов культивирования, будут представлены по крайней мере некоторыми небольшими сегментами последовательности.[13]
Секвенирование с высокой пропускной способностью
Преимущество высокопроизводительного секвенирования заключается в том, что этот метод не требует клонирования ДНК перед секвенированием, что устраняет одно из основных предубеждений и узких мест при отборе проб окружающей среды. Первые метагеномные исследования, проведенные с использованием высокопроизводительное секвенирование используется массово параллельно 454 пиросеквенирование.[17] Три другие технологии, обычно применяемые для отбора проб окружающей среды: Персональная геномная машина Ion Torrent, то Иллюмина MiSeq или HiSeq и Прикладные биосистемы SOLiD система.[22] Эти методы секвенирования ДНК генерируют более короткие фрагменты, чем Секвенирование по Сэнгеру; Система Ion Torrent PGM и пиросеквенирование 454 обычно производят чтения ~ 400 пар оснований, Illumina MiSeq производит считывания 400-700 пар оснований (в зависимости от того, используются ли параметры парных концов), а SOLiD производит считывания 25-75 пар оснований.[23] Исторически сложилось так, что эти длины чтения были значительно короче, чем типичная длина чтения секвенирования Сэнгера, составляющая ~ 750 п.н., однако технология Illumina быстро приближается к этому эталону. Однако это ограничение компенсируется гораздо большим количеством считываний последовательности. В 2009 году пиросеквенированные метагеномы генерируют 200–500 мегабаз, а платформы Illumina генерируют около 20–50 гигабаз, но за последние годы эти объемы выросли на порядки.[24]
Новый подход сочетает в себе последовательность действий при дробовике и захват конформации хромосомы (Hi-C), который измеряет близость любых двух последовательностей ДНК в одной и той же клетке, чтобы направлять сборку микробного генома.[25] Технологии долгого секвенирования, включая PacBio RSII и PacBio Sequel от Тихоокеанские биологические науки, и Nanopore MinION, Grigion, PrometION от Oxford Nanopore Technologies, - еще один вариант получения длинных считываний секвенирования ружья, которые должны упростить процесс сборки.[26]
Биоинформатика
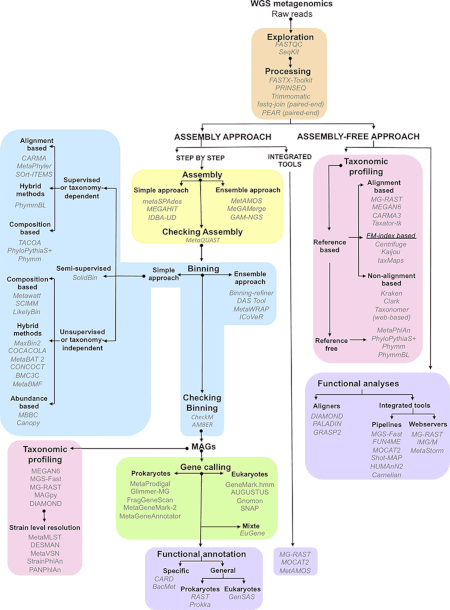
Данные, полученные в ходе метагеномических экспериментов, огромны и изначально зашумлены, они содержат фрагментированные данные, представляющие до 10 000 видов.[1] Последовательность коровы рубец метагеном сгенерирован 279 гигабазы, или 279 миллиардов пар оснований данных нуклеотидной последовательности,[28] в то время как человеческий кишечник микробиом Каталог генов выявил 3,3 миллиона генов, собранных из 567,7 гигабаз данных о последовательностях.[29] Сбор, обработка и извлечение полезной биологической информации из наборов данных такого размера представляют собой серьезные вычислительные задачи для исследователей.[21][30][31][32]
Предварительная фильтрация последовательности
Первый шаг анализа метагеномных данных требует выполнения определенных шагов предварительной фильтрации, включая удаление повторяющихся низкокачественных последовательностей и последовательностей вероятных эукариотический происхождения (особенно в метагеномах человеческого происхождения).[33][34] Доступные методы удаления загрязняющих последовательностей геномной ДНК эукариот включают Eu-Detect и DeConseq.[35][36]
сборка
Данные последовательностей ДНК из геномных и метагеномных проектов по существу одинаковы, но данные геномных последовательностей предлагают более высокие покрытие в то время как метагеномные данные обычно не являются избыточными.[31] Кроме того, более широкое использование технологий секвенирования второго поколения с короткими длинами считывания означает, что большая часть будущих метагеномных данных будет подвержена ошибкам. В совокупности эти факторы делают сборку считываемых метагеномных последовательностей в геномы трудными и ненадежными. Неправильная сборка вызвана наличием повторяющиеся последовательности ДНК которые делают сборку особенно сложной из-за разницы в относительной численности видов, присутствующих в выборке.[37] Неправильная сборка может также включать сочетание последовательностей более чем одного вида в химерные контиги.[37]
Существует несколько программ сборки, большинство из которых может использовать информацию из парные конечные теги с целью повышения точности сборок. Некоторые программы, например Phrap или же Celera Assembler, были разработаны для сборки одиночных геномы но тем не менее дают хорошие результаты при сборке наборов метагеномных данных.[1] Другие программы, например Бархатный сборщик, были оптимизированы для более коротких чтений, произведенных секвенированием второго поколения за счет использования графы де Брейна.[38][39] Использование эталонных геномов позволяет исследователям улучшить сборку наиболее распространенных видов микробов, но этот подход ограничен небольшим набором микробных типов, для которых доступны секвенированные геномы.[37] После создания сборки возникает дополнительная задача - «метагеномная деконволюция» или определение того, какие последовательности происходят от каких видов в образце.[40]
Прогноз генов
Метагеномный анализ трубопроводы использовать два подхода к аннотации областей кодирования в собранных контигах.[37] Первый подход - выявить гены на основе гомология с генами, которые уже общедоступны в базы данных последовательностей обычно ВЗРЫВ поиски. Такой подход реализован в программе. МЕГАН 4.[41] Второй, ab initio, использует внутренние особенности последовательности для прогнозирования кодирующих областей на основе обучающих наборов генов из родственных организмов. Это подход, используемый такими программами, как GeneMark[42] и Мерцание. Главное преимущество ab initio предсказание состоит в том, что он позволяет обнаруживать кодирующие области, у которых отсутствуют гомологи в базах данных последовательностей; однако он наиболее точен, когда для сравнения доступны большие участки смежной геномной ДНК.[1]
Видовое разнообразие
Аннотации генов предоставляют «что», а измерения видовое разнообразие укажите «кто».[43] Чтобы связать состав сообщества и функцию в метагеномах, последовательности должны быть объединены. Биннинг это процесс связывания определенной последовательности с организмом.[37] В биннинге на основе подобия такие методы, как ВЗРЫВ используются для быстрого поиска филогенетических маркеров или других подобных последовательностей в существующих общедоступных базах данных. Такой подход реализован в МЕГАН.[44] Другой инструмент, PhymmBL, использует интерполированные марковские модели назначить чтения.[1] МетаФлан и АМФОРА представляют собой методы, основанные на уникальных маркерах, специфичных для клады, для оценки относительной численности организмов с улучшенными вычислительными характеристиками.[45] Другие инструменты, например МОТ[46][47] и MetaPhyler,[48] использовать универсальные гены-маркеры для профилирования видов прокариот. С Профайлер mOTUs можно профилировать виды без эталонного генома, улучшая оценку разнообразия микробного сообщества.[47] Недавние методы, такие как SLIMM используйте ландшафт охвата считыванием отдельных эталонных геномов, чтобы свести к минимуму ложноположительные совпадения и получить надежные относительные количества.[49] В бининге на основе композиции в методах используются внутренние характеристики последовательности, такие как частоты олигонуклеотидов или систематическая ошибка использования кодонов.[1] После объединения последовательностей можно проводить сравнительный анализ разнообразия и богатства.
Интеграция данных
Огромный объем экспоненциально растущих данных последовательностей представляет собой серьезную проблему, которая осложняется сложностью метаданные связанные с метагеномными проектами. Метаданные включают подробную информацию о трехмерном (включая глубину или высоту) географическом положении и особенностях окружающей среды образца, физические данные о месте отбора проб и методологию отбора образцов.[31] Эта информация необходима как для обеспечения воспроизводимость и включить последующий анализ. Из-за своей важности метаданные и совместный обзор и обработка данных требуют стандартизованных форматов данных, размещенных в специализированных базах данных, таких как Genomes OnLine Database (GOLD).[50]
Было разработано несколько инструментов для интеграции метаданных и данных о последовательностях, что позволяет проводить последующий сравнительный анализ различных наборов данных с использованием ряда экологических индексов. В 2007 году Фолкер Мейер и Роберт Эдвардс и команда компании Аргоннская национальная лаборатория и Чикагский университет выпустила быструю аннотацию Metagenomics с использованием сервера Subsystem Technology (МГ-РАСТ ) ресурс сообщества для анализа набора данных метагенома.[51] По состоянию на июнь 2012 г. более 14,8 терабаз (14x1012 баз) ДНК были проанализированы с более чем 10 000 общедоступных наборов данных, свободно доступных для сравнения в MG-RAST. Более 8000 пользователей отправили в MG-RAST в общей сложности 50 000 метагеномов. В Интегрированные микробные геномы / метагеномы (IMG / M) система также предоставляет набор инструментов для функционального анализа микробных сообществ на основе их метагеномной последовательности, основанной на геномах эталонных изолятов, включенных из Интегрированные микробные геномы (IMG) и Геномная энциклопедия бактерий и архей (ГЭБА) проект.[52]
Одним из первых автономных инструментов для анализа высокопроизводительных метагеномных данных о дробовике был МЕГАН (Анализатор генома MEta).[41][44] Первая версия программы была использована в 2005 году для анализа метагеномного контекста последовательностей ДНК, полученных из кости мамонта.[17] Основываясь на сравнении BLAST со справочной базой данных, этот инструмент выполняет как таксономическое, так и функциональное объединение, помещая считывания в узлы таксономии NCBI с использованием простого алгоритма наименьшего общего предка (LCA) или на узлы СЕМЯ или же КЕГГ классификации соответственно.[53]
С появлением быстрых и недорогих инструментов для секвенирования рост баз данных последовательностей ДНК сейчас экспоненциальный (например, база данных NCBI GenBank [54]). Необходимы более быстрые и эффективные инструменты, чтобы идти в ногу с высокопроизводительным секвенированием, поскольку подходы на основе BLAST, такие как MG-RAST или MEGAN, работают медленно для аннотирования больших выборок (например, несколько часов для обработки набора данных / выборки небольшого / среднего размера [55]). Таким образом, недавно появились сверхбыстрые классификаторы благодаря более доступным мощным серверам. Эти инструменты могут выполнять таксономическую аннотацию с чрезвычайно высокой скоростью, например CLARK [56] (согласно авторам CLARK, он может точно классифицировать «32 миллиона метагеномных коротких чтений в минуту»). При такой скорости очень большой набор данных / выборка из миллиарда коротких чтений может быть обработан примерно за 30 минут.
С увеличением доступности образцов, содержащих древнюю ДНК, и из-за неопределенности, связанной с природой этих образцов (повреждение древней ДНК),[57] Появился быстрый инструмент, позволяющий производить консервативные оценки сходства. По словам авторов FALCON, он может использовать ослабленные пороги и редактировать расстояния без ущерба для памяти и скорости.
Сравнительная метагеномика
Сравнительный анализ метагеномов может дать дополнительную информацию о функции сложных микробных сообществ и их роли в здоровье хозяина.[58] Попарные или множественные сравнения между метагеномами могут производиться на уровне композиции последовательности (сравнение GC-контент или размер генома), таксономическое разнообразие или функциональное дополнение. Сравнение популяционной структуры и филогенетического разнообразия может проводиться на основе 16S и других филогенетических маркерных генов или - в случае сообществ с низким разнообразием - путем реконструкции генома из набора метагеномных данных.[59] Функциональные сравнения между метагеномами могут быть выполнены путем сравнения последовательностей со справочными базами данных, такими как COG или же КЕГГ и табулирование численности по категориям и оценка любых различий на предмет статистической значимости.[53] Этот геноцентрический подход подчеркивает функциональное дополнение сообщество в целом, а не таксономические группы, и показывает, что функциональные дополнения аналогичны в аналогичных условиях окружающей среды.[59] Следовательно, метаданные об экологическом контексте метагеномной выборки особенно важны для сравнительного анализа, поскольку они предоставляют исследователям возможность изучать влияние среды обитания на структуру и функции сообщества.[1]
Кроме того, в нескольких исследованиях также использовались шаблоны использования олигонуклеотидов для выявления различий между различными микробными сообществами. Примеры таких методологий включают подход относительного содержания динуклеотидов, разработанный Willner et al.[60] и подход HabiSign Ghosh et al.[61] Это последнее исследование также показало, что различия в моделях использования тетрануклеотидов могут быть использованы для идентификации генов (или метагеномных считываний), происходящих из определенных мест обитания. Дополнительно некоторые методы, такие как TriageTools[62] или Compareads[63] обнаруживать похожие чтения между двумя наборами чтения. В мера сходства они применяются при чтении основаны на количестве одинаковых слов длины k разделяется парами чтений.
Ключевой целью сравнительной метагеномики является определение группы (групп) микробов, которые отвечают за придание определенных характеристик данной среде. Однако из-за проблем, связанных с технологиями секвенирования, артефакты необходимо учитывать, как и в случае с metagenomeSeq.[30] Другие охарактеризовали межмикробные взаимодействия между резидентными микробными группами. А GUI приложение для сравнительного метагеномного анализа под названием Community-Analyzer было разработано Kuntal et al. [64] который реализует алгоритм компоновки графиков на основе корреляции, который не только облегчает быструю визуализацию различий в анализируемых микробных сообществах (с точки зрения их таксономического состава), но также дает представление о внутренних межмикробных взаимодействиях, происходящих в них. Примечательно, что этот алгоритм компоновки также позволяет группировать метагеномы на основе вероятных паттернов межмикробного взаимодействия, а не просто сравнивать значения численности различных таксономических групп. Кроме того, инструмент реализует несколько интерактивных функций на основе графического интерфейса пользователя, которые позволяют пользователям выполнять стандартный сравнительный анализ микробиомов.
Анализ данных
Обмен веществ в сообществе
Во многих бактериальных сообществах, естественных или искусственно созданных (например, биореакторы ), наблюдается значительное разделение труда в метаболизме (Синтрофия ), во время которого продукты жизнедеятельности одних организмов становятся метаболитами для других.[65] В одной из таких систем метаногенный биореактор, функциональная стабильность требует наличия нескольких синтрофический разновидность (Syntrophobacterales и Синергия ) работая вместе, чтобы превратить сырые ресурсы в полностью метаболизированные отходы (метан ).[66] Используя сравнительные исследования генов и эксперименты по экспрессии с микрочипы или же протеомика исследователи могут собрать воедино метаболическую сеть, выходящую за пределы видовых границ. Такие исследования требуют детальных знаний о том, какие версии каких белков кодируются какими видами и даже какими штаммами каких видов. Следовательно, геномная информация сообщества - еще один фундаментальный инструмент (с метаболомика и протеомика) в стремлении определить, как метаболиты передаются и трансформируются сообществом.[67]
Метатранскриптомика
Метагеномика позволяет исследователям получить доступ к функциональному и метаболическому разнообразию микробных сообществ, но не может показать, какие из этих процессов являются активными.[59] Извлечение и анализ метагеномных мРНК (в метатранскриптом) предоставляет информацию о регулирование и выражение профили сложных сообществ. Из-за технических трудностей ( короткий период полураспада мРНК, например) в коллекции РНК окружающей среды было относительно мало на месте метатранскриптомические исследования микробных сообществ на сегодняшний день.[59] Хотя изначально ограничено микрочип технологии, исследования метатранскриптомики использовали технологии транскриптомики для измерения экспрессии всего генома и количественной оценки микробного сообщества,[59] впервые применен при анализе окисления аммиака в почвах.[68]
Вирусы
Метагеномное секвенирование особенно полезно при изучении вирусных сообществ. Поскольку у вирусов нет общего универсального филогенетического маркера (как 16S РНК для бактерий и архей, и 18S РНК for eukarya), единственный способ получить доступ к генетическому разнообразию вирусного сообщества из образца окружающей среды - это метагеномика. Таким образом, вирусные метагеномы (также называемые виромами) должны предоставлять все больше и больше информации о вирусном разнообразии и эволюции.[69][70][71][72][73] Например, метагеномный конвейер под названием Гигантский поиск вирусов показал первые доказательства существования гигантские вирусы в солёной пустыне[74] и в засушливых антарктических долинах.[75]
Приложения
Метагеномика может способствовать развитию знаний в самых разных областях. Его также можно применять для решения практических задач в лекарство, инженерное дело, сельское хозяйство, устойчивость и экология.[31][76]
сельское хозяйство
В почвы в которых растут растения, населяют микробные сообщества, причем в одном грамме почвы содержится около 109-1010 микробные клетки, которые содержат примерно одну гигабазу информации о последовательности.[77][78] Микробные сообщества, населяющие почвы, являются одними из самых сложных, известных науке, и остаются малоизученными, несмотря на их экономическое значение.[79] Микробные консорциумы выполняют самые разнообразные экосистемные услуги необходим для роста растений, в том числе для фиксации атмосферного азота, круговорот питательных веществ, подавление болезней и секвестр утюг и другие металлы.[80] Стратегии функциональной метагеномики используются для изучения взаимодействия между растениями и микробами посредством независимого от культивирования исследования этих микробных сообществ.[81][82] Позволяя понять роль ранее некультурных или редких членов сообщества в круговороте питательных веществ и стимулировании роста растений, метагеномные подходы могут способствовать более эффективному выявлению заболеваний в посевы и домашний скот и адаптация усиленного сельское хозяйство методы, которые улучшают здоровье сельскохозяйственных культур, используя взаимоотношения между микробами и растениями.[31]
Биотопливо
Биотопливо находятся топливо происходит от биомасса преобразование, как в преобразовании целлюлоза содержалась в кукуруза стебли просо, и другую биомассу в целлюлозный этанол.[31] Этот процесс зависит от микробных консорциумов (ассоциаций), которые превращают целлюлозу в сахара, за которым следует ферментация сахаров в этиловый спирт. Микробы также производят различные источники биоэнергетика включая метан и водород.[31]
В эффективная деконструкция в промышленном масштабе биомассы требует нового ферменты с более высокой производительностью и более низкой стоимостью.[28] Метагеномные подходы к анализу сложных микробных сообществ позволяют целенаправленно скрининг из ферменты с промышленным применением в производстве биотоплива, например гликозидгидролазы.[83] Кроме того, знание того, как эти микробные сообщества функционируют, необходимо для их контроля, а метагеномика является ключевым инструментом в их понимании. Метагеномные подходы позволяют проводить сравнительный анализ между сходящийся микробные системы, такие как биогаз ферментеры[84] или же насекомое травоядные животные такой как грибной сад из муравьи-листорезы.[85]
Биотехнологии
Сообщества микроорганизмов производят широкий спектр биологически активных химикатов, которые используются в конкуренции и общении.[80] Многие из используемых сегодня лекарств изначально были обнаружены в микробах; Недавний прогресс в разработке богатого генетического ресурса некультивируемых микробов привел к открытию новых генов, ферментов и натуральных продуктов.[59][86] Применение метагеномики позволило развить товар и тонкие химикаты, агрохимикаты и фармацевтические препараты где выгода катализируемый ферментами хиральный синтез получает все большее признание.[87]
В программе используются два типа анализа. биоразведка метагеномных данных: функционально-управляемый скрининг выраженного признака и управляемый последовательностью скрининг интересующих последовательностей ДНК.[88] Функционально-управляемый анализ направлен на идентификацию клонов, экспрессирующих желаемый признак или полезную активность, с последующей биохимической характеристикой и анализом последовательности. Этот подход ограничен доступностью подходящего скрининга и требованием, чтобы желаемый признак был выражен в клетке-хозяине. Более того, низкий уровень обнаружения (менее одного на 1000 проверенных клонов) и его трудоемкость еще больше ограничивают этот подход.[89] Напротив, анализ на основе последовательностей использует консервативные последовательности ДНК к разработка праймеров для ПЦР для скрининга клонов на интересующую последовательность.[88] По сравнению с подходами, основанными на клонировании, использование подхода, основанного только на последовательностях, дополнительно сокращает объем требуемой лабораторной работы. Применение массового параллельного секвенирования также значительно увеличивает объем генерируемых данных о последовательностях, что требует высокопроизводительных конвейеров биоинформатического анализа.[89] Подход к скринингу, основанный на последовательностях, ограничен широтой и точностью функций генов, представленных в общедоступных базах данных последовательностей. На практике в экспериментах используется комбинация как функционального, так и последовательного подходов, основанных на интересующей функции, сложности выборки, подлежащей скринингу, и других факторах.[89][90] Пример успеха использования метагеномики в качестве биотехнологии для открытия лекарств проиллюстрирован на примере малацидин антибиотики.[91]
Экология

Метагеномика может дать ценную информацию о функциональной экологии экологических сообществ.[92] Метагеномный анализ бактериальных консорциумов, обнаруженных в испражнениях австралийских морских львов, предполагает, что богатые питательными веществами фекалии морских львов могут быть важным источником питательных веществ для прибрежных экосистем. Это связано с тем, что бактерии, которые выделяются одновременно с дефекацией, способны расщеплять питательные вещества с фекалиями до биодоступной формы, которая может быть использована в пищевой цепи.[93]
Секвенирование ДНК также можно использовать в более широком смысле для идентификации видов, присутствующих в водоеме,[94] мусор, отфильтрованный из воздуха, или образец грязи. Это может установить диапазон инвазивные виды и вымирающие виды и отслеживать сезонные популяции.
Восстановление окружающей среды
Метагеномика может улучшить стратегии мониторинга воздействия загрязняющие вещества на экосистемы и для очистки загрязненной окружающей среды. Более глубокое понимание того, как микробные сообщества справляются с загрязнителями, улучшает оценки потенциала загрязненных участков для восстановления после загрязнения и увеличивает шансы биоаугментация или же биостимуляция испытания, чтобы добиться успеха.[95]
Характеристика кишечных микробов
Микробные сообщества играют ключевую роль в сохранении человеческих здоровье, но их состав и механизм, с помощью которого они это делают, остаются загадочными.[96] Метагеномное секвенирование используется для характеристики микробных сообществ из 15–18 участков тела не менее 250 человек. Это часть Инициатива человеческого микробиома с основными целями, чтобы определить, есть ли ядро человеческий микробиом, чтобы понять изменения в микробиоме человека, которые могут быть связаны со здоровьем человека, и разработать новые технологические и биоинформатика инструменты для поддержки этих целей.[97]
Другое медицинское исследование в рамках проекта MetaHit (Метагеномика кишечного тракта человека) состояло из 124 человек из Дании и Испании, состоящих из здоровых пациентов с избыточным весом и с заболеваниями раздраженного кишечника. В исследовании была сделана попытка классифицировать глубину и филогенетическое разнообразие желудочно-кишечных бактерий. Используя данные последовательности Illumina GA и SOAPdenovo, инструмент на основе графов де Брейна, специально разработанный для коротких чтений сборки, они смогли сгенерировать 6,58 миллионов контигов размером более 500 пар оснований для общей длины контигов 10,3 ГБ и длины N50 2,2 kb.
Исследование показало, что два бактериальных подразделения, Bacteroidetes и Firmicutes, составляют более 90% известных филогенетических категорий, которые доминируют над бактериями дистальных отделов кишечника. Используя относительные частоты генов, обнаруженные в кишечнике, эти исследователи идентифицировали 1244 метагеномных кластера, которые критически важны для здоровья кишечного тракта. В этих группах диапазонов есть два типа функций: домашнее хозяйство и функции кишечника. Кластеры генов домашнего хозяйства необходимы всем бактериям и часто являются основными участниками основных метаболических путей, включая центральный углеродный метаболизм и синтез аминокислот. Специфические для кишечника функции включают адгезию к белкам хозяина и сбор сахаров из глобосерийных гликолипидов. Было показано, что пациенты с синдромом раздраженного кишечника имеют на 25% меньше генов и более низкое бактериальное разнообразие, чем люди, не страдающие синдромом раздраженного кишечника, что указывает на то, что изменения в разнообразии биомов кишечника пациентов могут быть связаны с этим состоянием.
Хотя эти исследования подчеркивают некоторые потенциально ценные медицинские приложения, только 31–48,8% считываний можно сопоставить с 194 общедоступными бактериальными геномами кишечника человека и 7,6–21,2% - с геномами бактерий, доступными в GenBank, что указывает на то, что для захватить новые бактериальные геномы.[98]
Диагностика инфекционных заболеваний
Различение инфекционных и неинфекционных заболеваний и определение основной этиологии инфекции может быть довольно сложной задачей. Например, более половины случаев энцефалит остаются недиагностированными, несмотря на обширное тестирование с использованием современных клинических лабораторных методов. Метагеномное секвенирование перспективно как чувствительный и быстрый метод диагностики инфекции путем сравнения генетического материала, обнаруженного в образце пациента, с базой данных, содержащей тысячи бактерий, вирусов и других патогенов.
Смотрите также
Рекомендации
- ^ а б c d е ж грамм Вули Дж. К., Годзик А., Фридберг И. (февраль 2010 г.). Bourne PE (ред.). «Букварь по метагеномике». PLOS вычислительная биология. 6 (2): e1000667. Bibcode:2010PLSCB ... 6E0667W. Дои:10.1371 / journal.pcbi.1000667. ЧВК 2829047. PMID 20195499.
- ^ а б Hugenholtz P, Goebel BM, Pace NR (сентябрь 1998 г.). «Влияние культурно-независимых исследований на формирующееся филогенетическое представление о бактериальном разнообразии». Журнал бактериологии. 180 (18): 4765–74. Дои:10.1128 / JB.180.18.4765-4774.1998. ЧВК 107498. PMID 9733676.
- ^ Марко, Д., изд. (2011). Метагеномика: современные инновации и будущие тенденции. Caister Academic Press. ISBN 978-1-904455-87-5.
- ^ Эйзен Дж. А. (март 2007 г.). «Экологическое секвенирование ружья: его потенциал и проблемы для изучения скрытого мира микробов». PLOS Биология. 5 (3): e82. Дои:10.1371 / journal.pbio.0050082. ЧВК 1821061. PMID 17355177.
- ^ Хандельсман Дж., Рондон М.Р., Брэди С.Ф., Кларди Дж., Гудман Р.М. (октябрь 1998 г.). «Молекулярно-биологический доступ к химии неизвестных почвенных микробов: новый рубеж для натуральных продуктов». Химия и биология. 5 (10): R245-9. Дои:10.1016 / S1074-5521 (98) 90108-9. PMID 9818143..
- ^ Чен К., Пахтер Л. (июль 2005 г.). «Биоинформатика для полногеномного секвенирования микробных сообществ». PLOS вычислительная биология. 1 (2): 106–12. Bibcode:2005PLSCB ... 1 ... 24C. Дои:10.1371 / journal.pcbi.0010024. ЧВК 1185649. PMID 16110337.
- ^ Lane DJ, Pace B, Olsen GJ, Stahl DA, Sogin ML, Pace NR (октябрь 1985 г.). «Быстрое определение последовательностей рибосомной РНК 16S для филогенетических анализов». Труды Национальной академии наук Соединенных Штатов Америки. 82 (20): 6955–9. Bibcode:1985PNAS ... 82.6955L. Дои:10.1073 / pnas.82.20.6955. ЧВК 391288. PMID 2413450.
- ^ Пейс Н. Р., Шталь Д. А., Лейн Д. Д., Олсен Г. Дж. (1986). «Анализ природных микробных популяций по последовательностям рибосомных РНК». В Marshall KC (ред.). Успехи в микробной экологии. 9. Springer США. С. 1–55. Дои:10.1007/978-1-4757-0611-6_1. ISBN 978-1-4757-0611-6.
- ^ Шмидт Т.М., Делонг Е.Ф., Пейс Н.Р. (июль 1991 г.). «Анализ морского сообщества пикопланктона путем клонирования и секвенирования гена 16S рРНК». Журнал бактериологии. 173 (14): 4371–8. Дои:10.1128 / jb.173.14.4371-4378.1991. ЧВК 208098. PMID 2066334.
- ^ Хили Ф.Г., Рэй Р.М., Олдрич Х.С., Уилки А.С., Инграм Л.О., Шанмугам К.Т. (1995). «Прямое выделение функциональных генов, кодирующих целлюлазы, из микробного консорциума в термофильном анаэробном варочном котле, поддерживаемом лигноцеллюлозой». Прикладная микробиология и биотехнология. 43 (4): 667–74. Дои:10.1007 / BF00164771. PMID 7546604. S2CID 31384119.
- ^ Штейн Дж. Л., Марш Т. Л., Ву К. Ю., Шизуя Х., Делонг Е. Ф. (февраль 1996 г.). «Характеристика некультивируемых прокариот: выделение и анализ фрагмента генома с парой 40 килобаз из планктонного морского архея». Журнал бактериологии. 178 (3): 591–9. Дои:10.1128 / jb.178.3.591-599.1996. ЧВК 177699. PMID 8550487.
- ^ Брейтбарт М., Саламон П., Андресен Б., Махаффи Дж. М., Сегалл А. М., Мид Д. и др. (Октябрь 2002 г.). «Геномный анализ некультивируемых морских вирусных сообществ». Труды Национальной академии наук Соединенных Штатов Америки. 99 (22): 14250–5. Bibcode:2002PNAS ... 9914250B. Дои:10.1073 / pnas.202488399. ЧВК 137870. PMID 12384570.
- ^ а б Тайсон Г.В., Чепмен Дж., Хугенгольц П., Аллен Э., Рам Р. Дж., Ричардсон П. М. и др. (Март 2004 г.). «Структура сообщества и метаболизм через реконструкцию микробных геномов из окружающей среды». Природа. 428 (6978): 37–43. Bibcode:2004 Натур 428 ... 37 т. Дои:10.1038 / природа02340. PMID 14961025. S2CID 4420754.(требуется подписка)
- ^ Гугенгольц П. (2002). «Изучение прокариотического разнообразия в эпоху генома». Геномная биология. 3 (2): ОБЗОРЫ0003. Дои:10.1186 / gb-2002-3-2-reviews0003. ЧВК 139013. PMID 11864374.
- ^ Вентер Дж. К., Ремингтон К., Гейдельберг Дж. Ф., Халперн А. Л., Руш Д., Эйзен Дж. А. и др. (Апрель 2004 г.). «Секвенирование экологического генома Саргассова моря». Наука. 304 (5667): 66–74. Bibcode:2004Наука ... 304 ... 66В. CiteSeerX 10.1.1.124.1840. Дои:10.1126 / science.1093857. PMID 15001713. S2CID 1454587.
- ^ Юзеф С., Нилсон К. Х., Руш Д. Б., МакКроу Дж. П., Дюпон С. Л., Ким М. и др. (Ноябрь 2010 г.). «Геномная и функциональная адаптация планктонных прокариот над поверхностью океана». Природа. 468 (7320): 60–6. Bibcode:2010Натура.468 ... 60л. Дои:10.1038 / природа09530. PMID 21048761.(требуется подписка)
- ^ а б c Пойнар Х.Н., Шварц С., Ци Дж., Шапиро Б., Макфи Р.Д., Буиг Б. и др. (Январь 2006 г.). «От метагеномики к палеогеномике: масштабное секвенирование ДНК мамонта». Наука. 311 (5759): 392–4. Bibcode:2006Научный ... 311..392P. Дои:10.1126 / science.1123360. PMID 16368896. S2CID 11238470.
- ^ Эдвардс Р.А., Родригес-Брито Б., Вегли Л., Хейнс М., Брейтбарт М., Петерсон Д.М. и др. (Март 2006 г.). «Использование пиросеквенирования, чтобы пролить свет на микробную экологию глубоких шахт». BMC Genomics. 7: 57. Дои:10.1186/1471-2164-7-57. ЧВК 1483832. PMID 16549033.
- ^ Томас Т., Гилберт Дж, Мейер Ф (февраль 2012 г.). «Метагеномика - руководство от выборки до анализа данных». Микробная информатика и эксперименты. 2 (1): 3. Дои:10.1186/2042-5783-2-3. ЧВК 3351745. PMID 22587947.
- ^ Бежа О, Сузуки М.Т., Кунин Э.В., Аравинд Л., Хадд А., Нгуен Л.П. и др. (Октябрь 2000 г.). «Создание и анализ бактериальных библиотек искусственных хромосом из набора морских микробов». Экологическая микробиология. 2 (5): 516–29. Дои:10.1046 / j.1462-2920.2000.00133.x. PMID 11233160. S2CID 8267748.
- ^ а б Сегата Н., Берниген Д., Щекотка Т.Л., Морган XC, Гарретт WS, Хаттенхауэр С. (май 2013 г.). «Вычислительная метаомика для исследований микробного сообщества». Молекулярная системная биология. 9 (666): 666. Дои:10.1038 / msb.2013.22. ЧВК 4039370. PMID 23670539.
- ^ Родриг С., Матерна А.С., Тимберлейк СК, Блэкберн М.К., Мальмстрем Р.Р., Альм Э.Дж., Чисхолм ЮЗ (июль 2010 г.). Гилберт JA (ред.). «Разблокировка короткого секвенирования чтения для метагеномики». PLOS ONE. 5 (7): e11840. Bibcode:2010PLoSO ... 511840R. Дои:10.1371 / journal.pone.0011840. ЧВК 2911387. PMID 20676378.
- ^ Schuster SC (январь 2008 г.). «Секвенирование следующего поколения меняет сегодняшнюю биологию». Методы природы. 5 (1): 16–8. Дои:10.1038 / nmeth1156. PMID 18165802. S2CID 1465786.
- ^ «Метагеномика против закона Мура». Методы природы. 6 (9): 623. 2009. Дои:10.1038 / nmeth0909-623.
- ^ Стюарт Р.Д., Аффрет, доктор медицины, Варр А., Вайзер А.Х., Пресс МО, Лэнгфорд К.В. и др. (Февраль 2018). «Сборка 913 микробных геномов из метагеномного секвенирования рубца коровы». Nature Communications. 9 (1): 870. Bibcode:2018НатКо ... 9..870С. Дои:10.1038 / s41467-018-03317-6. ЧВК 5830445. PMID 29491419.
- ^ Хираока С., Ян СС, Ивасаки В. (сентябрь 2016 г.). «Метагеномика и биоинформатика в микробной экологии: текущее состояние и перспективы». Микробы и окружающая среда. 31 (3): 204–12. Дои:10.1264 / jsme2.ME16024. ЧВК 5017796. PMID 27383682.
- ^ Перес-Кобас А.Е., Гомес-Валеро Л., Бухризер С. (2020). «Метагеномные подходы в микробной экологии: обновленная информация об анализе секвенирования полногеномных и маркерных генов». Микробная геномика. 6 (8). Дои:10.1099 / мген.0.000409. PMID 32706331.
- ^ а б Hess M, Sczyrba A, Egan R, Kim TW, Chokhawala H, Schroth G и др. (Январь 2011 г.). «Метагеномное открытие генов и геномов, разлагающих биомассу, из рубца коров». Наука. 331 (6016): 463–7. Bibcode:2011Научный ... 331..463H. Дои:10.1126 / science.1200387. PMID 21273488. S2CID 36572885.
- ^ Цинь Дж., Ли Р., Раес Дж., Арумугам М., Бургдорф К.С., Маничан С. и др. (Март 2010 г.). «Каталог микробных генов кишечника человека, созданный с помощью метагеномного секвенирования». Природа. 464 (7285): 59–65. Bibcode:2010Натура 464 ... 59.. Дои:10.1038 / природа08821. ЧВК 3779803. PMID 20203603.(требуется подписка)
- ^ а б Полсон JN, Stine OC, Bravo HC, Pop M (декабрь 2013 г.). «Дифференциальный анализ численности для исследований микробных маркеров генов». Методы природы. 10 (12): 1200–2. Дои:10.1038 / nmeth.2658. ЧВК 4010126. PMID 24076764.
- ^ а б c d е ж грамм Комитет по метагеномике: проблемы и функциональные применения, Национальный исследовательский совет (2007). Новая наука метагеномики: раскрытие секретов нашей микробной планеты. Вашингтон, округ Колумбия: The National Academies Press. Дои:10.17226/11902. ISBN 978-0-309-10676-4. PMID 21678629.
- ^ Улас А., Павлоуди С., Полименаку П., Павлопулос Г.А., Папаниколау Н., Котулас Г. и др. (2015). «Метагеномика: инструменты и идеи для анализа данных секвенирования следующего поколения, полученных из исследований биоразнообразия». Биоинформатика и биология. 9: 75–88. Дои:10.4137 / BBI.S12462. ЧВК 4426941. PMID 25983555.
- ^ Mende DR, Waller AS, Sunagawa S, Järvelin AI, Chan MM, Arumugam M, et al. (23 февраля 2012 г.). «Оценка метагеномной сборки с использованием смоделированных данных секвенирования следующего поколения». PLOS ONE. 7 (2): e31386. Bibcode:2012PLoSO ... 731386M. Дои:10.1371 / journal.pone.0031386. ЧВК 3285633. PMID 22384016.
- ^ Balzer S, Malde K, Grohme MA, Jonassen I (апрель 2013 г.). «Фильтрация повторяющихся чтений из 454 данных пиросеквенирования». Биоинформатика. 29 (7): 830–6. Дои:10.1093 / биоинформатика / btt047. ЧВК 3605598. PMID 23376350.
- ^ Мохаммед М.Х., Чадарам С., Командури Д., Гош Т.С., Манде СС (сентябрь 2011 г.). «Eu-Detect: алгоритм для обнаружения эукариотических последовательностей в наборах метагеномных данных». Журнал биологических наук. 36 (4): 709–17. Дои:10.1007 / s12038-011-9105-2. PMID 21857117. S2CID 25857874.
- ^ Шмидер Р., Эдвардс Р. (март 2011 г.). «Быстрая идентификация и устранение контаминации последовательностей из наборов геномных и метагеномных данных». PLOS ONE. 6 (3): e17288. Bibcode:2011PLoSO ... 617288S. Дои:10.1371 / journal.pone.0017288. ЧВК 3052304. PMID 21408061.
- ^ а б c d е Кунин В., Коупленд А., Лапидус А., Мавроматис К., Гугенгольц П. (декабрь 2008 г.). «Справочник биоинформатика по метагеномике». Обзоры микробиологии и молекулярной биологии. 72 (4): 557–78, Содержание. Дои:10.1128 / MMBR.00009-08. ЧВК 2593568. PMID 19052320.
- ^ Намики Т., Хачия Т., Танака Х., Сакакибара Ю. (ноябрь 2012 г.). «MetaVelvet: расширение ассемблера Velvet для сборки метагенома de novo из коротких последовательностей чтения». Исследования нуклеиновых кислот. 40 (20): e155. Дои:10.1093 / нар / gks678. ЧВК 3488206. PMID 22821567.
- ^ Зербино Д.Р., Бирни Э. (май 2008 г.). "Velvet: алгоритмы сборки короткого чтения de novo с использованием графов де Брейна". Геномные исследования. 18 (5): 821–9. Дои:10.1101 / гр.074492.107. ЧВК 2336801. PMID 18349386.
- ^ Бертон Дж., Лячко И., Данэм М. Дж., Шендур Дж. (Май 2014 г.). "Деконволюция на уровне видов метагеномных сборок с помощью карт вероятности контакта на основе Hi-C". G3. 4 (7): 1339–46. Дои:10.1534 / g3.114.011825. ЧВК 4455782. PMID 24855317.
- ^ а б Huson DH, Mitra S, Ruscheweyh HJ, Weber N, Schuster SC (сентябрь 2011 г.). «Интегративный анализ экологических последовательностей с использованием MEGAN4». Геномные исследования. 21 (9): 1552–60. Дои:10.1101 / гр.120618.111. ЧВК 3166839. PMID 21690186.
- ^ Жу В., Ломсадзе А., Бородовский М. (июль 2010 г.). «Идентификация гена Ab initio в метагеномных последовательностях». Исследования нуклеиновых кислот. 38 (12): e132. Дои:10.1093 / nar / gkq275. ЧВК 2896542. PMID 20403810.
- ^ Конопка А (ноябрь 2009 г.). «Что такое экология микробного сообщества?». Журнал ISME. 3 (11): 1223–30. Дои:10.1038 / ismej.2009.88. PMID 19657372.
- ^ а б Huson DH, Auch AF, Qi J, Schuster SC (март 2007 г.). «МЕГАН анализ метагеномных данных». Геномные исследования. 17 (3): 377–86. Дои:10.1101 / гр.5969107. ЧВК 1800929. PMID 17255551.
- ^ Сегата Н., Уолдрон Л., Балларини А., Нарасимхан В., Юссон О., Хаттенхауэр С. (июнь 2012 г.). «Профилирование метагеномного микробного сообщества с использованием уникальных маркерных генов, специфичных для клады». Методы природы. 9 (8): 811–4. Дои:10.1038 / nmeth.2066. ЧВК 3443552. PMID 22688413.
- ^ Sunagawa S, Mende DR, Zeller G, Izquierdo-Carrasco F, Berger SA, Kultima JR и др. (Декабрь 2013). «Метагеномное профилирование видов с использованием универсальных филогенетических маркерных генов». Методы природы. 10 (12): 1196–9. Дои:10.1038 / nmeth.2693. PMID 24141494. S2CID 7728395.
- ^ а б Milanese A, Mende DR, Paoli L, Salazar G, Ruscheweyh HJ, Cuenca M и др. (Март 2019 г.). «Численность, активность и геномное профилирование популяций с помощью mOTU2». Nature Communications. 10 (1): 1014. Bibcode:2019НатКо..10.1014M. Дои:10.1038 / s41467-019-08844-4. ЧВК 6399450. PMID 30833550.
- ^ Лю Б., Гиббонс Т., Годси М., Треанген Т., Поп М. (2011). «Точная и быстрая оценка таксономических профилей по метагеномным последовательностям дробовика». BMC Genomics. 12 Приложение 2: S4. Дои:10.1186 / 1471-2164-12-S2-S4. ЧВК 3194235. PMID 21989143.
- ^ Дади TH, Renard BY, Wieler LH, Semmler T, Reinert K (2017). «SLIMM: идентификация микроорганизмов из метагеномов на видовом уровне». PeerJ. 5: e3138. Дои:10.7717 / peerj.3138. ЧВК 5372838. PMID 28367376.
- ^ Пагани И., Лиолиос К., Янссон Дж., Чен И.М., Смирнова Т., Носрат Б. и др. (Январь 2012 г.). «База данных Genomes OnLine (GOLD) v.4: статус геномных и метагеномных проектов и связанных с ними метаданных». Исследования нуклеиновых кислот. 40 (Проблема с базой данных): D571-9. Дои:10.1093 / nar / gkr1100. ЧВК 3245063. PMID 22135293.
- ^ Мейер Ф., Паарманн Д., Д'Суза М., Олсон Р., Гласс Э.М., Кубал М. и др. (Сентябрь 2008 г.). «RAST-сервер метагеномики - общедоступный ресурс для автоматического филогенетического и функционального анализа метагеномов». BMC Bioinformatics. 9: 386. Дои:10.1186/1471-2105-9-386. ЧВК 2563014. PMID 18803844.
- ^ Марковиц В.М., Чен И.М., Чу К., Сзето Э., Паланиаппан К., Гречкин Ю. и др. (Январь 2012 г.). «IMG / M: интегрированная система управления данными метагенома и сравнительного анализа». Исследования нуклеиновых кислот. 40 (Проблема с базой данных): D123-9. Дои:10.1093 / nar / gkr975. ЧВК 3245048. PMID 22086953.
- ^ а б Митра С., Рупек П., Рихтер Д.К., Урих Т., Гилберт Дж. А., Мейер Ф. и др. (Февраль 2011 г.). «Функциональный анализ метагеномов и метатранскриптомов с использованием SEED и KEGG». BMC Bioinformatics. 12 Приложение 1: S21. Дои:10.1186 / 1471-2105-12-S1-S21. ЧВК 3044276. PMID 21342551.
- ^ Бенсон Д.А., Кавано М., Кларк К., Карш-Мизрахи И., Липман Д.И., Остелл Дж., Сэйерс Е.В. (январь 2013 г.). «ГенБанк». Исследования нуклеиновых кислот. 41 (Проблема с базой данных): D36-42. Дои:10.1093 / нар / gks1195. ЧВК 3531190. PMID 23193287.
- ^ Базине А.Л., Каммингс МП (май 2012 г.). «Сравнительная оценка программ классификации последовательностей». BMC Bioinformatics. 13: 92. Дои:10.1186/1471-2105-13-92. ЧВК 3428669. PMID 22574964.
- ^ Ounit R, Wanamaker S, Close TJ, Lonardi S (март 2015 г.). «CLARK: быстрая и точная классификация метагеномных и геномных последовательностей с использованием дискриминационных k-мер». BMC Genomics. 16: 236. Дои:10.1186 / s12864-015-1419-2. ЧВК 4428112. PMID 25879410.
- ^ Пратас Д., Пинхо А.Дж., Сильва Р.М., Родригес Дж.М., Хоссейни М., Каэтано Т., Феррейра П.Дж. (февраль 2018 г.). «СОКОЛ: метод определения метагеномного состава древней ДНК». bioRxiv 10.1101/267179.
- ^ Курокава К., Ито Т., Кувахара Т., Осима К., Тох Х, Тойода А. и др. (Август 2007 г.). «Сравнительная метагеномика выявила обычно обогащенные наборы генов в микробиомах кишечника человека». ДНК исследования. 14 (4): 169–81. Дои:10.1093 / днарес / dsm018. ЧВК 2533590. PMID 17916580.
- ^ а б c d е ж Саймон С., Дэниел Р. (февраль 2011 г.). «Метагеномный анализ: прошлые и будущие тенденции». Прикладная и экологическая микробиология. 77 (4): 1153–61. Дои:10.1128 / AEM.02345-10. ЧВК 3067235. PMID 21169428.
- ^ Виллнер Д., Тербер Р.В., Ровер Ф. (июль 2009 г.). «Метагеномные сигнатуры 86 микробных и вирусных метагеномов». Экологическая микробиология. 11 (7): 1752–66. Дои:10.1111 / j.1462-2920.2009.01901.x. PMID 19302541.
- ^ Гош Т.С., Мохаммед М.Х., Раджасинг Х., Чадарам С., Манде С.С. (2011). «HabiSign: новый подход к сравнению метагеномов и быстрой идентификации последовательностей, специфичных для среды обитания». BMC Bioinformatics. 12 Дополнение 13 (Дополнение 13): S9. Дои:10.1186 / 1471-2105-12-s13-s9. ЧВК 3278849. PMID 22373355.
- ^ Фимерели Д., Объездные В., Конопка Т. (апрель 2013 г.). «TriageTools: инструменты для разделения и приоритезации анализа данных высокопроизводительного секвенирования». Исследования нуклеиновых кислот. 41 (7): e86. Дои:10.1093 / nar / gkt094. ЧВК 3627586. PMID 23408855.
- ^ Maillet N, Lemaitre C, Chikhi R, Lavenier D, Peterlongo P (2012). "Compareads: сравнение огромных метагеномных экспериментов". BMC Bioinformatics. 13 Дополнение 19 (Дополнение 19): S10. Дои:10.1186 / 1471-2105-13-S19-S10. ЧВК 3526429. PMID 23282463.
- ^ Кунтал Б.К., Гош Т.С., Манде СС (октябрь 2013 г.). «Сообщество-анализатор: платформа для визуализации и сравнения структуры микробного сообщества по микробиомам». Геномика. 102 (4): 409–18. Дои:10.1016 / j.ygeno.2013.08.004. PMID 23978768.
- ^ Вернер Дж. Дж., Найтс Д., Гарсия М. Л., Скальфон Н. Б., Смит С., Ярашески К. и др. (Март 2011 г.). «Структуры бактериальных сообществ уникальны и устойчивы в полномасштабных биоэнергетических системах». Труды Национальной академии наук Соединенных Штатов Америки. 108 (10): 4158–63. Bibcode:2011PNAS..108.4158W. Дои:10.1073 / pnas.1015676108. ЧВК 3053989. PMID 21368115.
- ^ Макинерни MJ, Sieber JR, Gunsalus RP (декабрь 2009 г.). «Синтрофия в анаэробных глобальных углеродных циклах». Текущее мнение в области биотехнологии. 20 (6): 623–32. Дои:10.1016 / j.copbio.2009.10.001. ЧВК 2790021. PMID 19897353.
- ^ Klitgord N, Segrè D (август 2011 г.). «Экосистемная биология микробного метаболизма». Текущее мнение в области биотехнологии. 22 (4): 541–6. Дои:10.1016 / j.copbio.2011.04.018. PMID 21592777.
- ^ Лейнингер С., Урих Т., Шлотер М., Шварк Л., Ци Дж., Николь Г.В. и др. (Август 2006 г.). «Среди прокариот, окисляющих аммиак, в почвах преобладают археи». Природа. 442 (7104): 806–9. Bibcode:2006 Натур.442..806L. Дои:10.1038 / природа04983. PMID 16915287. S2CID 4380804.
- ^ Паез-Эспино Д., Элоэ-Фадрош Е.А., Павлопулос Г.А., Томас А.Д., Хантеманн М., Михайлова Н. и др. (Август 2016 г.). «Открытие вирома Земли». Природа. 536 (7617): 425–30. Bibcode:2016Натура.536..425P. Дои:10.1038 / природа19094. PMID 27533034. S2CID 4466854.
- ^ Паез-Эспино Д., Чен И.А., Паланиаппан К., Ратнер А., Чу К., Сзето Э. и др. (Январь 2017 г.). «IMG / VR: база данных культивируемых и некультивируемых ДНК-вирусов и ретровирусов». Исследования нуклеиновых кислот. 45 (D1): D457 – D465. Дои:10.1093 / нар / gkw1030. ЧВК 5210529. PMID 27799466.
- ^ Паез-Эспино Д., Ру С., Чен И.А., Паланиаппан К., Ратнер А., Чу К. и др. (Январь 2019). «IMG / VR v.2.0: интегрированная система управления и анализа данных для культивируемых и экологических вирусных геномов». Исследования нуклеиновых кислот. 47 (D1): D678 – D686. Дои:10.1093 / нар / gky1127. ЧВК 6323928. PMID 30407573.
- ^ Паез-Эспино Д., Павлопулос Г.А., Иванова Н.Н., Кирпидес NC (август 2017 г.). «Конвейер обнаружения ненаправленных вирусных последовательностей и кластеризация вирусов для метагеномных данных» (PDF). Протоколы природы. 12 (8): 1673–1682. Дои:10.1038 / nprot.2017.063. PMID 28749930. S2CID 2127494.
- ^ Кристенсен Д.М., Мушегян А.Р., Доля В.В., Кунин Е.В. (январь 2010 г.). «Новые измерения мира вирусов открыты с помощью метагеномики». Тенденции в микробиологии. 18 (1): 11–9. Дои:10.1016 / j.tim.2009.11.003. ЧВК 3293453. PMID 19942437.
- ^ Kerepesi C, Grolmusz V (март 2016 г.). «Гигантские вирусы пустыни Кач». Архив вирусологии. 161 (3): 721–4. arXiv:1410.1278. Дои:10.1007 / s00705-015-2720-8. PMID 26666442. S2CID 13145926.
- ^ Kerepesi C, Grolmusz V (июнь 2017 г.). «Поиск гигантских вирусов» обнаруживает изобилие гигантских вирусов в засушливых долинах Антарктики ». Архив вирусологии. 162 (6): 1671–1676. arXiv:1503.05575. Дои:10.1007 / s00705-017-3286-4. PMID 28247094. S2CID 1925728.
- ^ Copeland CS (сентябрь – октябрь 2017 г.). «Мир внутри нас» (PDF). Журнал здравоохранения Нового Орлеана: 21–26.
- ^ Янссон Дж. (2011). "Навстречу" Тера-Терра ": терабазное секвенирование наземных метагеномов Печать E-mail". Микроб. 6 (7). п. 309. Архивировано с оригинал 31 марта 2012 г.
- ^ Vogel TM, Simonet P, Jansson JK, Hirsch PR, Tiedje JM, Van Elsas JD, Bailey MJ, Nalin R, Philippot L (2009). «TerraGenome: консорциум для секвенирования метагенома почвы». Обзоры природы Микробиология. 7 (4): 252. Дои:10.1038 / nrmicro2119.
- ^ "Домашняя страница TerraGenome". Международный консорциум по секвенированию TerraGenome. Получено 30 декабря 2011.
- ^ а б Комитет по метагеномике: проблемы и функциональное применение, Национальный исследовательский совет (2007). Понимание нашей микробной планеты: новая наука метагеномики (PDF). Издательство национальных академий.
- ^ Чарльз Т. (2010). «Возможности исследования взаимодействия растений и микробов с использованием методов метагеномики». Метагеномика: теория, методы и приложения. Caister Academic Press. ISBN 978-1-904455-54-7.
- ^ Брингель Ф., Couée I (22 мая 2015 г.). «Основные роли микроорганизмов филлосферы на стыке между функционированием растений и динамикой газовых примесей в атмосфере». Границы микробиологии. 6: 486. Дои:10.3389 / fmicb.2015.00486. ЧВК 4440916. PMID 26052316.
- ^ Ли Л.Л., Маккоркл С.Р., Мончи С., Тагави С., ван дер Лели Д. (май 2009 г.). «Биоразведка метагеномов: гликозилгидролазы для преобразования биомассы». Биотехнология для биотоплива. 2: 10. Дои:10.1186/1754-6834-2-10. ЧВК 2694162. PMID 19450243.
- ^ Янике С., Андер С., Бекель Т., Бисдорф Р., Дрёге М., Гартеман К. Х. и др. (Январь 2011 г.). Азиз РК (ред.). «Сравнительный и совместный анализ двух наборов метагеномных данных из ферментера биогаза, полученных с помощью 454-пиросеквенирования». PLOS ONE. 6 (1): e14519. Bibcode:2011PLoSO ... 614519J. Дои:10.1371 / journal.pone.0014519. ЧВК 3027613. PMID 21297863.
- ^ Суен Дж., Скотт Дж. Дж., Эйлвард Ф. О., Адамс С. М., Триндж С. Г., Пинто-Томас А. А. и др.(Сентябрь 2010 г.). Зонненбург J (ред.). «Микробиом насекомых-травоядных животных с высокой способностью разлагать биомассу растений». PLOS Genetics. 6 (9): e1001129. Дои:10.1371 / journal.pgen.1001129. ЧВК 2944797. PMID 20885794.
- ^ Саймон С., Дэниел Р. (ноябрь 2009 г.). «Достижения и новые знания, полученные с помощью метагеномных подходов». Прикладная микробиология и биотехнология. 85 (2): 265–76. Дои:10.1007 / s00253-009-2233-z. ЧВК 2773367. PMID 19760178.
- ^ Вонг Д. (2010). «Применение метагеномики для промышленных биопродуктов». Метагеномика: теория, методы и приложения. Caister Academic Press. ISBN 978-1-904455-54-7.
- ^ а б Schloss PD, Handelsman J (июнь 2003 г.). «Биотехнологические перспективы от метагеномики» (PDF). Текущее мнение в области биотехнологии. 14 (3): 303–10. Дои:10.1016 / S0958-1669 (03) 00067-3. PMID 12849784. Архивировано из оригинал (PDF) 4 марта 2016 г.. Получено 20 января 2012.
- ^ а б c Kakirde KS, Parsley LC, Liles MR (ноябрь 2010 г.). «Размер имеет значение: подходы к метагеномике почвы, основанные на применении». Биология и биохимия почвы. 42 (11): 1911–1923. Дои:10.1016 / j.soilbio.2010.07.021. ЧВК 2976544. PMID 21076656.
- ^ Парачин Н.С., Горва-Грауслунд М.Ф. (май 2011 г.). «Выделение изомераз ксилозы путем скрининга на основе последовательности и функции из почвенной метагеномной библиотеки». Биотехнология для биотоплива. 4 (1): 9. Дои:10.1186/1754-6834-4-9. ЧВК 3113934. PMID 21545702.
- ^ Hover BM, Kim SH, Katz M, Charlop-Powers Z, Owen JG, Ternei MA и др. (Апрель 2018). «Независимое от культуры открытие малацидинов как кальций-зависимых антибиотиков с активностью против грамположительных патогенов с множественной лекарственной устойчивостью». Природная микробиология. 3 (4): 415–422. Дои:10.1038 / s41564-018-0110-1. ЧВК 5874163. PMID 29434326.
- ^ Raes J, Letunic I, Yamada T, Jensen LJ, Bork P (март 2011 г.). «К экологии, основанной на молекулярных признаках, путем интеграции биогеохимических, географических и метагеномных данных». Молекулярная системная биология. 7: 473. Дои:10.1038 / msb.2011.6. ЧВК 3094067. PMID 21407210.
- ^ Лавери Т.Дж., Роуднью Б., Сеймур Дж., Митчелл Дж. Г., Джеффрис Т. (2012). Стейнке Д. (ред.). «В микробном метагеноме фекалий австралийского морского льва (Neophoca cinerea) обнаружен высокий потенциал транспорта и циркуляции питательных веществ». PLOS ONE. 7 (5): e36478. Bibcode:2012PLoSO ... 736478L. Дои:10.1371 / journal.pone.0036478. ЧВК 3350522. PMID 22606263.
- ^ «Что плавает в реке? Просто поищите ДНК». NPR.org. 24 июля 2013 г.. Получено 10 октября 2014.
- ^ Георгий I, Стенуит Б., Агатос С.Н. (2010). «Применение метагеномики для биоремедиации». В Марко Д. (ред.). Метагеномика: теория, методы и приложения. Caister Academic Press. ISBN 978-1-904455-54-7.
- ^ Циммер С. (13 июля 2010 г.). «Как микробы защищают и определяют нас». Нью-Йорк Таймс. Получено 29 декабря 2011.
- ^ Нельсон К.Э. и Уайт Б.А. (2010). «Метагеномика и ее приложения к изучению микробиома человека». Метагеномика: теория, методы и приложения. Caister Academic Press. ISBN 978-1-904455-54-7.
- ^ Цинь Дж., Ли Р., Раес Дж., Арумугам М., Бургдорф К.С., Маничан С. и др. (Март 2010 г.). «Каталог микробных генов кишечника человека, созданный с помощью метагеномного секвенирования». Природа. 464 (7285): 59–65. Bibcode:2010Натура 464 ... 59.. Дои:10.1038 / природа08821. ЧВК 3779803. PMID 20203603.
внешняя ссылка
- Сосредоточьтесь на метагеномике в Обзоры природы Микробиология сайт журнала
- Инициатива «Критическая оценка интерпретации метагенома» (CAMI) оценить методы метагеномики
| Геномика | |
|---|---|
| Биоинформатика | |
| Структурная биология | |
| Инструменты исследования | |
| Организации |
|
| |