Канцерогенез - Carcinogenesis

Канцерогенез, также называется онкогенез или туморогенез, это формирование рак, при этом нормальный клетки находятся преобразованный в раковые клетки. Процесс характеризуется изменениями на клеточном, генетический, и эпигенетический уровни и аномальные деление клеток. Деление клеток - это физиологический процесс, который происходит практически во всех ткани и при различных обстоятельствах. Обычно баланс между пролиферацией и запрограммированной гибелью клеток в виде апоптоз, поддерживается для обеспечения целостности тканей и органы. Согласно общепринятой теории канцерогенеза, теория соматических мутаций, мутации в ДНК и эпимутации которые приводят к раку, нарушают эти упорядоченные процессы, нарушая программирование, регулирующее процессы, нарушая нормальный баланс между пролиферацией и гибелью клеток. Это приводит к неконтролируемому делению клеток и эволюция этих клеток от естественный отбор в организме. Только определенные мутации приводят к раку, тогда как большинство мутаций - нет.
Варианты наследственных генов могут предрасполагать людей к раку. Кроме того, факторы окружающей среды, такие как канцерогены и радиация вызывают мутации, которые могут способствовать развитию рака. Наконец, случайные ошибки в нормальной репликации ДНК могут привести к мутациям, вызывающим рак.[1] Обычно требуется серия из нескольких мутаций определенных классов генов, прежде чем нормальная клетка превратится в раковая клетка.[2][3][4][5] В среднем, например, при раке толстой кишки обнаруживается 15 «водительских мутаций» и 60 «пассажирских» мутаций.[2] Мутации в генах, регулирующих деление клеток, апоптоз (смерть клетки), и Ремонт ДНК может привести к неконтролируемой пролиферации клеток и раку.
Рак по сути, это болезнь регуляции роста тканей. Чтобы нормальная клетка преобразовать в раковую клетку, гены которые регулируют рост и дифференцировку клеток, должны быть изменены.[6] Генетические и эпигенетические изменения могут происходить на многих уровнях, от приобретения или потери целых хромосом до мутации, затрагивающей одиночный нуклеотид ДНК или к подавлению или активации микроРНК, которая контролирует экспрессию от 100 до 500 генов.[7][8] Эти изменения затрагивают две широкие категории генов. Онкогены могут быть нормальные гены, которые экспрессируются на чрезмерно высоких уровнях, или измененные гены, обладающие новыми свойствами. В любом случае экспрессия этих генов способствует злокачественному фенотипу раковых клеток. Гены-супрессоры опухолей представляют собой гены, которые подавляют деление клеток, выживание или другие свойства раковых клеток. Гены-супрессоры опухолей часто отключаются из-за генетических изменений, способствующих развитию рака. в заключение Oncovirinae, вирусы которые содержат онкоген, классифицируются как онкогенные, потому что они вызывают рост опухолевых тканей в хозяин. Этот процесс также называют вирусная трансформация.
Причины
Генетические и эпигенетические
Существует разнообразная схема классификации различных геномных изменений, которые могут способствовать генерации раковые клетки. Многие из этих изменений мутации, или изменения в нуклеотид последовательность геномной ДНК. Также существует множество эпигенетических изменений, которые влияют на то, экспрессируются гены или нет. Анеуплоидия, наличие ненормального количества хромосом, это одно изменение генома, которое не является мутацией, и может включать в себя приобретение или потерю одного или нескольких хромосомы через ошибки в митоз. Крупномасштабные мутации включают удаление или усиление части хромосомы. Геномная амплификация происходит, когда клетка набирает много копий (часто 20 или более) небольшой хромосомной области, обычно содержащей один или несколько онкогенов и прилегающий генетический материал. Перемещение возникает, когда две отдельные хромосомные области неправильно сливаются, часто в характерном месте. Хорошо известным примером этого является Филадельфийская хромосома, или транслокация хромосом 9 и 22, которая происходит в хронический миелолейкоз, и приводит к созданию BCR -abl гибридный белок, онкогенный тирозинкиназа. Мелкомасштабные мутации включают: точечные мутации, удаления, и вставки, что может произойти в промоутер гена и повлиять на его выражение, или может встречаться в генах кодирующая последовательность и изменить функцию или стабильность его белок товар. Нарушение одного гена также может быть результатом интеграция геномного материала из ДНК-вирус или ретровирус, и такое событие может также привести к экспрессии вирусных онкогенов в пораженной клетке и ее потомках.
Повреждение ДНК

Повреждение ДНК считается основной причиной рака.[9] Более 60000 новых естественных случаев повреждения ДНК возникают в среднем на одну человеческую клетку в день из-за эндогенных клеточных процессов (см. Статью Повреждение ДНК (естественное) ).
Дополнительное повреждение ДНК может возникнуть в результате воздействия экзогенный агенты. В качестве одного из примеров экзогенный канцерогенный агент, табачный дым вызывает повышенное повреждение ДНК, и это повреждение ДНК, вероятно, вызывает рост рака легких из-за курения.[10] В других примерах УФ-свет от солнечного излучения вызывает повреждение ДНК, что важно для меланома,[11] Helicobacter pylori инфекция вызывает высокий уровень активные формы кислорода которые повреждают ДНК и способствуют рак желудка,[12] и Aspergillus flavus метаболит афлатоксин представляет собой агент, повреждающий ДНК, вызывающий рак печени.[13]
Повреждение ДНК также может быть вызвано вещества, вырабатываемые в организме. Макрофаги и нейтрофилы в воспаленном эпителии толстой кишки являются источником активных форм кислорода, вызывающих повреждение ДНК, которое инициирует туморогенез,[14] и желчные кислоты, присутствующие в большом количестве в толстой кишке людей, придерживающихся диеты с высоким содержанием жиров, также вызывают повреждение ДНК и способствуют развитию рака толстой кишки.[15]
Такие экзогенные и эндогенные источники повреждения ДНК указаны в прямоугольниках вверху рисунка в этом разделе. Центральная роль повреждения ДНК в прогрессировании рака показана на втором уровне рисунка. Центральные элементы повреждения ДНК, эпигенетический Изменения и недостаточная репарация ДНК при прогрессировании рака показаны красным.
Дефицит репарации ДНК приведет к накоплению большего количества повреждений ДНК и увеличит риск рака. Например, люди с наследственным нарушением в любой из 34 Гены репарации ДНК (см. статью Расстройство дефицита репарации ДНК ) подвержены повышенному риску рака, при этом некоторые дефекты вызывают до 100% пожизненной вероятности рака (например, p53 мутации).[16] Такие мутации зародышевой линии показаны в рамке слева от рисунка с указанием их вклада в дефицит репарации ДНК. Однако такие мутации зародышевой линии (которые вызывают высокую пенетрант онкологические синдромы) являются причиной только примерно один процент рака.[17]
Большинство видов рака называют ненаследственными или «спорадическими». Около 30% спорадических видов рака действительно имеют наследственный компонент, который в настоящее время не определен, в то время как большинство, или 70% спорадических видов рака, не имеют наследственного компонента.[18]
При спорадическом раке дефицит репарации ДНК иногда возникает из-за мутации в гене репарации ДНК; гораздо чаще снижение или отсутствие экспрессии генов репарации ДНК происходит из-за эпигенетические изменения которые уменьшают или молчание экспрессии гена. Это показано на рисунке на 3-м уровне сверху. Например, из 113 обследованных последовательно опухолей прямой кишки только четыре имели миссенс-мутация в гене репарации ДНК MGMT, в то время как у большинства из них экспрессия MGMT снижена из-за метилирование МГМТ промоутер регион (эпигенетическое изменение).[19]
Когда экспрессия генов репарации ДНК снижается, это вызывает дефицит репарации ДНК. Это показано на рисунке на 4-м уровне сверху. При дефиците репарации ДНК повреждение ДНК сохраняется в клетках на более высоком, чем обычно, уровне (5-й уровень сверху на рисунке); это избыточное повреждение вызывает повышенную частоту мутаций и / или эпимутация (6-й уровень сверху рисунка). Экспериментально частота мутаций значительно увеличивается в клетках, дефектных по Ремонт несоответствия ДНК[20][21] или в Гомологичный рекомбинационный ремонт (HRR).[22] Хромосомные перестройки и анеуплоидия также увеличение количества HRR-дефектных клеток[23] Во время репарации двухцепочечных разрывов ДНК или репарации других повреждений ДНК не полностью очищенные участки репарации могут вызывать эпигенетическое молчание генов.[24][25]
Соматические мутации и эпигенетические изменения, вызванные повреждением ДНК и недостаточностью репарации ДНК, накапливаются в полевые дефекты. Полевые дефекты представляют собой нормально выглядящие ткани с множественными изменениями (обсуждаются в разделе ниже) и являются обычными предшественниками развития неупорядоченного и чрезмерно пролиферирующего клона ткани при раке. Такие дефекты поля (второй уровень снизу рисунка) могут иметь многочисленные мутации и эпигенетические изменения.
Невозможно определить первоначальную причину большинства конкретных видов рака. В некоторых случаях существует только одна причина: например, вирус HHV-8 вызывает все Саркомы Капоши. Однако с помощью эпидемиология рака методы и информацию, можно произвести оценку вероятной причины во многих других ситуациях. Например, рак легких имеет несколько причин, включая употребление табака и радон. Мужчины, которые в настоящее время курят табак, заболевают раком легких в 14 раз чаще, чем мужчины, которые никогда не курили табак: вероятность рака легких у курильщика, вызванного курением, составляет около 93%; с вероятностью 7% рак легких у курильщика был вызван газом радоном или другой причиной, не связанной с курением.[26] Эти статистические корреляции позволили исследователям сделать вывод о канцерогенности определенных веществ или поведения. Табачный дым вызывает повышенное экзогенный Повреждение ДНК, и это повреждение ДНК является вероятной причиной рака легких из-за курения. Среди более чем 5000 соединений табачного дыма генотоксичный Агенты, повреждающие ДНК, которые встречаются как в самых высоких концентрациях, так и оказывают сильнейшее мутагенное действие: акролеин, формальдегид, акрилонитрил, 1,3-бутадиен, ацетальдегид, окись этилена и изопрен.[10]
С помощью молекулярно-биологический методы, можно охарактеризовать мутации, эпимутации или хромосомные аберрации в опухоли, и стремительный прогресс наблюдается в области прогнозирования некоторых онкологических больных. прогноз на основе спектра мутаций. Например, до половины всех опухолей имеют дефектный ген p53. Эта мутация связана с плохим прогнозом, поскольку эти опухолевые клетки с меньшей вероятностью попадут в апоптоз или запрограммированная гибель клеток при повреждении терапией. Теломераза мутации устраняют дополнительные барьеры, увеличивая количество раз, которое клетка может делиться. Другие мутации позволяют опухоли вырастить новые кровеносные сосуды для обеспечения большего количества питательных веществ или для метастазировать, распространяясь на другие части тела. Однако, как только образуется рак, он продолжает развиваться и производить субклоны. В 2012 году сообщалось, что в одном образце рака почки, взятом в девяти разных областях, было 40 «повсеместных» мутаций, обнаруженных во всех девяти областях, 59 мутаций, общих для некоторых, но не всех девяти областей, и 29 «частных» мутаций только присутствует в одной области.[27]
Клоны клеток, в которых накапливаются все эти изменения ДНК, трудно проследить, но две недавние линии доказательств предполагают, что нормальные стволовые клетки могут быть клетками происхождения рака.[28][29] Во-первых, существует очень положительная корреляция (коэффициент Спирмена = 0,81; P <3,5 × 10-8) между риском развития рака в ткани и количеством нормальных делений стволовых клеток, происходящих в этой же ткани. Корреляция применялась к 31 типу рака и распространялась на пять порядки величины.[30] Эта корреляция означает, что если нормальные стволовые клетки из ткани делятся один раз, риск рака в этой ткани примерно в 1 раз. Если они разделятся в 1000 раз, риск рака увеличится в 1000 раз. И если нормальные стволовые клетки из ткани делятся 100000 раз, риск рака в этой ткани примерно в 100000 раз. Это убедительно свидетельствует о том, что основным фактором возникновения рака является тот факт, что «нормальные» стволовые клетки делятся, а это означает, что рак возникает в нормальных, здоровых стволовых клетках.[29]
Во-вторых, статистика показывает, что большинство раковых заболеваний у человека диагностируется у пожилых людей. Возможное объяснение заключается в том, что рак возникает из-за того, что клетки со временем накапливают повреждения. ДНК - единственный клеточный компонент, который может накапливать повреждения на протяжении всей жизни, а стволовые клетки - единственные клетки, которые могут передавать ДНК от зиготы к клеткам на поздних этапах жизни. Другие клетки, полученные из стволовых клеток, не сохраняют ДНК с самого начала жизни, пока не произойдет возможный рак. Это означает, что большинство видов рака возникает из нормальных стволовых клеток.[28][29]
Вклад полевых дефектов

Период, термин "канцеризация поля "был впервые использован в 1953 году для описания области или" поля "эпителия, которое было обусловлено (в то время) в значительной степени неизвестными процессами, чтобы предрасполагать его к развитию рака.[31] С тех пор термины «канцеризация поля» и «дефект поля» используются для описания предраковых тканей, в которых вероятно возникновение новых видов рака.
Дефекты поля были идентифицированы в связи с раком и важны для прогрессирования рака.[32][33] Однако Рубин отметил, что[34] что «подавляющее большинство исследований рака было проведено на четко определенных опухолях in vivo или на отдельных неопластических очагах in vitro. Тем не менее, есть доказательства того, что более 80% соматических мутаций, обнаруженных в мутаторный фенотип колоректальные опухоли человека возникают до начала терминальной клональной экспансии ... "[35] Более половины соматических мутаций, выявленных в опухолях, произошли в предопухолевой фазе (в поле дефекта) во время роста явно нормальных клеток. Также можно было бы ожидать, что многие из эпигенетических изменений, присутствующих в опухолях, могли возникнуть в предопухолевых полевых дефектах.[36]
В толстой кишке дефект поля, вероятно, возникает в результате естественного отбора мутантной или эпигенетически измененной клетки среди стволовых клеток в основании одной из кишечные крипты на внутренней поверхности толстой кишки. Мутантная или эпигенетически измененная стволовая клетка может заменять другие близлежащие стволовые клетки путем естественного отбора. Это может вызвать образование патологического участка ткани. На рисунке в этом разделе представлена фотография только что резецированный и продольно открытый сегмент толстой кишки, показывающий рак толстой кишки и четыре полипа. Под фотографией представлена схематическая диаграмма того, как мог образоваться большой участок мутантных или эпигенетически измененных клеток, показанный на диаграмме большой областью желтого цвета. Внутри этого первого большого участка на диаграмме (большого клона клеток) может произойти вторая такая мутация или эпигенетическое изменение, так что данная стволовая клетка приобретает преимущество по сравнению со своими соседями, и эта измененная стволовая клетка может клонально расширяться, образуя вторичный патч или субклон в пределах исходного патча. На схеме это обозначено четырьмя меньшими участками разного цвета в большой желтой исходной области. Внутри этих новых участков (субклонов) процесс может повторяться несколько раз, на что указывают еще более мелкие участки внутри четырех вторичных участков (с все еще разными цветами на диаграмме), которые клонально расширяются, пока не появятся стволовые клетки, которые генерируют либо небольшие участки. полипы или злокачественное новообразование (рак). На фотографии видимый дефект поля в этом сегменте толстой кишки привел к образованию четырех полипов (с указанием размера полипов 6 мм, 5 мм и двух по 3 мм, а также рака размером около 3 см в самом длинном измерении). Эти новообразования также обозначены (на схеме под фото) 4 маленькими желто-коричневыми кружками (полипы) и более крупной красной областью (рак). Рак на фотографии возник в слепой кишке толстой кишки, где толстая кишка соединяется с тонкой кишкой (помечено) и где находится аппендикс (помечено). Жир на фото находится вне внешней стенки толстой кишки. В сегменте толстой кишки, показанном здесь, толстая кишка была разрезана вдоль, чтобы обнажить ее внутреннюю поверхность и показать рак и полипы, возникающие внутри внутренней эпителиальной выстилки толстой кишки.
Если общий процесс, в результате которого возникают спорадические виды рака толстой кишки, представляет собой формирование пре-неопластического клона, который распространяется естественным отбором с последующим образованием внутренних субклонов внутри исходного клона и суб-субклонов внутри них, тогда рак толстой кишки обычно должны быть связаны с полями нарастающей патологии, отражая последовательность предраковых событий, и им предшествовать их. Наиболее обширная область аномалии (крайняя желтая неправильная область на диаграмме) будет отражать самое раннее событие в формировании злокачественного новообразования.
При экспериментальной оценке специфических дефектов репарации ДНК при раке было показано, что многие специфические дефекты репарации ДНК также возникают в полевых дефектах, окружающих эти раковые образования. В таблице ниже приведены примеры, для которых было показано, что дефицит репарации ДНК при раке вызван эпигенетическим изменением, и несколько более низкие частоты, с которыми тот же самый вызванный эпигенетикой дефицит репарации ДНК был обнаружен в дефекте окружающего поля.
| Рак | Ген | Частота при раке | Частота дефекта поля | Справка |
|---|---|---|---|---|
| Колоректальный | MGMT | 46% | 34% | [37] |
| Колоректальный | MGMT | 47% | 11% | [38] |
| Колоректальный | MGMT | 70% | 60% | [39] |
| Колоректальный | MSH2 | 13% | 5% | [38] |
| Колоректальный | ERCC1 | 100% | 40% | [40] |
| Колоректальный | PMS2 | 88% | 50% | [40] |
| Колоректальный | XPF | 55% | 40% | [40] |
| Голова и шея | MGMT | 54% | 38% | [41] |
| Голова и шея | MLH1 | 33% | 25% | [42] |
| Голова и шея | MLH1 | 31% | 20% | [43] |
| Желудок | MGMT | 88% | 78% | [44] |
| Желудок | MLH1 | 73% | 20% | [45] |
| Пищевод | MLH1 | 77%–100% | 23%–79% | [46] |
Некоторые из небольших полипов в области дефекта поля, показанные на фотографии вскрытого сегмента толстой кишки, могут быть относительно доброкачественными новообразованиями. В исследовании 1996 года полипов размером менее 10 мм, обнаруженных во время колоноскопии с последующими повторными колоноскопиями в течение 3 лет, 25% не изменились в размере, 35% регрессировали или уменьшились в размере и 40% увеличились в размере.[47]
Нестабильность генома
Раки, как известно, проявляют нестабильность генома или «мутаторный фенотип».[48] ДНК, кодирующая белок в ядре, составляет около 1,5% от общей геномной ДНК.[49] Внутри этой кодирующей белок ДНК (называемой экзом ), средний рак молочной железы или толстой кишки может иметь от 60 до 70 мутаций, изменяющих белок, из которых около 3 или 4 могут быть «драйверными» мутациями, а остальные могут быть «пассажирскими» мутациями.[36] Однако среднее количество мутаций в последовательности ДНК во всем геноме (включая районы, не кодирующие белок ) в образце ткани рака молочной железы составляет около 20 000.[50] В среднем образце ткани меланомы (меланомы имеют более высокую экзом частота мутаций),[36]) общее количество мутаций в последовательности ДНК составляет около 80 000.[51] Эти высокие частоты мутаций в общих нуклеотидных последовательностях в раковых опухолях предполагают, что часто раннее изменение дефекта поля, приводящее к раку (например, желтая область на диаграмме в предыдущем разделе), является недостатком репарации ДНК. Обнаружены большие полевые дефекты, окружающие рак толстой кишки (примерно до 10 см с каждой стороны от рака).[40] часто иметь эпигенетические дефекты в двух или трех белках репарации ДНК (ERCC1, ERCC4 (XPF) и / или PMS2 ) на всей площади дефекта поля. Когда экспрессия генов репарации ДНК снижается, повреждения ДНК накапливаются в клетках с более высокой, чем обычно, скоростью, и это избыточное повреждение вызывает повышенную частоту мутаций и / или эпимутаций. Скорость мутации сильно увеличивается в клетках, дефектных по Ремонт несоответствия ДНК[20][21] или в гомологичный рекомбинационный ремонт (HRR).[22] Дефицит репарации ДНК сам по себе может привести к накоплению повреждений ДНК и стать причиной ошибок. транслезионный синтез некоторых поврежденных участков могут возникнуть мутации. Кроме того, неправильное восстановление накопленных повреждений ДНК может привести к эпимутациям. Эти новые мутации и / или эпимутации могут обеспечить пролиферативное преимущество, создавая дефект поля. Хотя мутации / эпимутации в генах репарации ДНК сами по себе не дают селективного преимущества, они могут переноситься в качестве пассажиров в клетках, когда клетка приобретает дополнительную мутацию / эпимутацию, которая действительно обеспечивает пролиферативное преимущество.
Неосновные теории
Существует ряд теорий канцерогенеза и лечения рака, которые выходят за рамки основного научного мнения из-за отсутствия научного обоснования, логики или доказательной базы. Эти теории могут использоваться для обоснования различных альтернативных методов лечения рака. Их следует отличать от тех теорий канцерогенеза, которые имеют логическую основу в рамках основной биологии рака и из которых могут быть сделаны гипотезы, проверяемые традиционным способом.
Однако несколько альтернативных теорий канцерогенеза основаны на научных данных и получают все большее признание. Некоторые исследователи считают, что рак может быть вызван анеуплоидия (числовые и структурные аномалии в хромосомах)[52] а не мутациями или эпимутациями. Рак также считается метаболическим заболеванием, при котором клеточный метаболизм кислорода отклоняется от пути, генерирующего энергию (окислительного фосфорилирования ) к пути, который порождает активные формы кислорода.[53] Это вызывает переключение энергии с окислительного фосфорилирования на аэробный гликолиз (Гипотеза Варбурга ), а накопление активные формы кислорода ведущий к окислительный стресс («Теория окислительного стресса рака»).[53]
Ряд авторов поставили под сомнение предположение о том, что рак возникает в результате последовательных случайных мутаций, как излишне упрощенное, предполагая вместо этого, что рак возникает в результате неспособности организма подавлять врожденную, запрограммированную тенденцию к пролиферации.[54] Родственная теория предполагает, что рак - это атавизм, эволюционный возврат к более ранней форме многоклеточная жизнь.[55] Гены, ответственные за неконтролируемый рост клеток и сотрудничество между раковые клетки очень похожи на те, которые позволили первым многоклеточным формам жизни сгруппироваться и процветать. Эти гены все еще существуют в геномах более сложных многоклеточные животные, такие как люди, хотя недавно развитые гены держат их под контролем. Когда новые управляющие гены по какой-либо причине выходят из строя, клетка может вернуться к своему более примитивному программированию и бесконтрольно воспроизводить. Теория является альтернативой представлению о том, что рак начинается с клеток-изгоев, которые эволюционируют в организме. Вместо этого они обладают фиксированным числом примитивных генов, которые постепенно активируются, что дает им конечную изменчивость.[56] Другая эволюционная теория возвращает корни рака к происхождению эукариот (ядерная) клетка массивным горизонтальный перенос генов, когда геномы заражающих вирусов были расщеплены (и, таким образом, аттенуированы) хозяином, но их фрагменты интегрировались в геном хозяина в качестве иммунной защиты. Таким образом, рак возникает, когда редкая соматическая мутация рекомбинирует такие фрагменты в функциональный драйвер пролиферации клеток.[57]
Биология раковых клеток
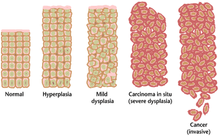
Часто множественные генетические изменения, приводящие к раку, накапливаются через много лет. В течение этого времени биологическое поведение предраковых клеток медленно меняется от свойств нормальных клеток к свойствам, подобным раковым. Предраковые ткани могут иметь отличительный внешний вид под микроскопом. Среди отличительных черт предзлокачественного поражения - повышенная количество делящихся клеток, изменение в ядерный размер и форма, изменение ячейки размер и форма, утрата специализированные функции ячейки, и потеря нормальной организации тканей. Дисплазия представляет собой аномальный тип чрезмерной пролиферации клеток, характеризующийся потерей нормального расположения тканей и клеточной структуры в предзлокачественных клетках. Эти ранние неопластический изменения следует отличать от гиперплазия, обратимое увеличение деления клеток, вызванное внешним раздражителем, например гормональным дисбалансом или хроническим раздражением.
Наиболее тяжелые случаи дисплазии называются карцинома in situ. На латыни термин на месте означает «на месте»; карцинома in situ относится к неконтролируемому росту диспластических клеток, который остается на своем первоначальном месте и не проявляется вторжение в другие ткани. Карцинома in situ может перерасти в инвазивную злокачественную опухоль и при обнаружении обычно удаляется хирургическим путем.
Клональная эволюция
Так же, как популяция животных подвергается эволюция, неконтролируемая популяция клеток также может претерпевать «эволюцию». Этот нежелательный процесс называется соматическая эволюция, и именно так возникает рак и со временем становится все более злокачественным.[58]
Большинство изменений клеточного метаболизма, которые позволяют клеткам беспорядочно расти, приводят к их гибели. Однако, как только начинается рак, раковые клетки пройти процесс естественный отбор: несколько клеток с новыми генетическими изменениями, которые увеличивают их выживаемость или воспроизводство, быстрее размножаются и вскоре начинают доминировать над растущей опухолью, поскольку клетки с менее благоприятными генетическими изменениями вытесняются.[59] Это тот же механизм, с помощью которого патогенный такие виды, как MRSA может стать устойчивый к антибиотикам и посредством чего ВИЧ может стать лекарственно устойчивый ), а также от болезней растений и насекомых. устойчивый к пестицидам. Эта эволюция объясняет, почему рак рецидив часто вовлекаются клетки, которые приобрели устойчивость к противораковым препаратам или устойчивость к радиации от лучевая терапия ).
Биологические свойства раковых клеток
В статье 2000 г. Ханахан и Вайнберг, биологические свойства клеток злокачественных опухолей были обобщены следующим образом:[60]
- Обретение самодостаточности в сигналы роста, что приводит к неконтролируемому росту.
- Потеря чувствительности к сигналам, препятствующим росту, что также приводит к неконтролируемому росту.
- Потеря емкости для апоптоз, позволяя расти, несмотря на генетические ошибки и внешние сигналы, препятствующие росту.
- Потеря емкости для старение, что приводит к безграничному репликативному потенциалу (бессмертию)
- Приобретение устойчивый ангиогенез, позволяя опухоли расти за пределы ограничений пассивной диффузии питательных веществ.
- Приобретение способности вторгаться в соседние ткани, определяющее свойство инвазивной карциномы.
- Приобретение способности сеять метастазы на удаленных участках - поздно появляющееся свойство некоторых злокачественных опухолей (карциномы и др.).
Выполнение этих нескольких шагов было бы очень редким событием без:
- Потеря способности исправлять генетические ошибки, что приводит к увеличению мутация скорость (геномная нестабильность), тем самым ускоряя все остальные изменения.
Эти биологические изменения являются классическими в карциномы; другие злокачественные опухоли, возможно, не потребуют их всех. Например, учитывая, что инвазия тканей и смещение к удаленным участкам являются нормальными свойствами лейкоциты, эти шаги не нужны при разработке лейкемия. Также разные этапы не обязательно представляют отдельные мутации. Например, инактивация одного гена, кодирующего p53 белок, вызовет геномную нестабильность, уклонение от апоптоза и усиление ангиогенеза. Кроме того, не все раковые клетки делятся. Скорее, подмножество клеток опухоли, называемое раковые стволовые клетки, реплицируются, поскольку они генерируют дифференцированные клетки.[61]
Рак как дефект клеточного взаимодействия
Обычно после повреждения или инфицирования ткани поврежденные клетки вызывают воспаление, стимулируя определенные паттерны активности ферментов и экспрессию генов цитокинов в окружающих клетках.[62][63] Выделяются отдельные кластеры («кластеры цитокинов») молекул, которые действуют как медиаторы, вызывая активность последующих каскадов биохимических изменений.[64] Каждый цитокин связывается со специфическими рецепторами на различных типах клеток, и каждый тип клеток в свою очередь реагирует, изменяя активность путей передачи внутриклеточного сигнала, в зависимости от рецепторов, которые клетка экспрессирует, и сигнальных молекул, присутствующих внутри клетки.[65][66] В совокупности этот процесс репрограммирования вызывает ступенчатое изменение фенотипов клеток, что в конечном итоге приведет к восстановлению функции ткани и к восстановлению существенной структурной целостности.[67][68] Таким образом, ткань может заживать в зависимости от продуктивной связи между клетками, присутствующими в месте повреждения, и иммунной системой.[69] Одним из ключевых факторов заживления является регуляция экспрессии генов цитокинов, которая позволяет комплементарным группам клеток реагировать на медиаторы воспаления способом, который постепенно вызывает существенные изменения в физиологии тканей.[70][71][72] Раковые клетки имеют в своем геноме постоянные (генетические) или обратимые (эпигенетические) изменения, которые частично препятствуют их взаимодействию с окружающими клетками и иммунной системой.[73][74] Раковые клетки не взаимодействуют со своим тканевым микроокружением таким образом, чтобы защитить целостность ткани; вместо этого движение и выживание раковых клеток становится возможным в тех местах, где они могут нарушать функцию тканей.[75][76] Раковые клетки выживают за счет «перенастройки» сигнальных путей, которые обычно защищают ткань от иммунной системы.
Одним из примеров перестройки функции ткани при раке является активность фактора транскрипции. NF-κB.[77]NF-κB активирует экспрессию многочисленных генов, участвующих в переходе от воспаления к регенерации, которые кодируют цитокины, факторы адгезии и другие молекулы, которые могут изменять судьбу клеток.[78] Такое перепрограммирование клеточных фенотипов обычно позволяет развиваться полностью функциональной неповрежденной ткани.[79] Активность NF-κB строго контролируется множеством белков, которые в совокупности гарантируют, что только дискретные кластеры генов индуцируются NF-κB в данной клетке и в данный момент времени.[80] Это жесткое регулирование обмена сигналами между клетками защищает ткань от чрезмерного воспаления и гарантирует, что различные типы клеток постепенно приобретают дополнительные функции и определенные положения. Нарушение этой взаимной регуляции между генетическим репрограммированием и клеточными взаимодействиями позволяет раковым клеткам вызывать метастазы. Раковые клетки аберрантно реагируют на цитокины и активируют сигнальные каскады, которые могут защитить их от иммунной системы.[77][81]
В рыбе
Роль йода в морской рыбе (богатой йодом) и пресноводной рыбе (йододефицитной) до конца не изучена, но сообщалось, что пресноводные рыбы более восприимчивы к инфекционным и, в частности, неопластическим и атеросклеротическим заболеваниям, чем морские. рыбы.[82][83] Морские двухстворчатые рыбы, такие как акулы, скаты и т. Д., В гораздо меньшей степени подвержены раку, чем пресноводные рыбы, и поэтому стимулировали медицинские исследования для лучшего понимания канцерогенеза.[84]
Механизмы
Чтобы клетки начали неконтролируемое деление, гены, регулирующие рост клеток, должны быть нарушены.[85] Протоонкогены гены, которые способствуют росту клеток и митоз, в то время как гены-супрессоры опухолей препятствовать росту клеток или временно останавливать деление клеток для выполнения Ремонт ДНК. Обычно серия из нескольких мутации к этим генам требуется до того, как нормальная клетка превратится в раковая клетка.[5] Это понятие иногда называют «онкоэволюцией». Мутации в этих генах дают сигнал опухолевым клеткам начать неконтролируемое деление. Но неконтролируемое деление клеток, которое характерно для рака, также требует, чтобы делящаяся клетка дублировала все свои клеточные компоненты для создания двух дочерних клеток. Активация анаэробного гликолиза ( Эффект варбурга ), что не обязательно вызвано мутациями в протоонкогенах и генах-супрессорах опухолей,[86] обеспечивает большинство строительных блоков, необходимых для дублирования клеточных компонентов делящейся клетки, и, следовательно, также важен для канцерогенеза.[53]
Онкогены
Онкогены способствовать росту клеток различными способами. Многие могут производить гормоны, «химический посланник» между клетками, который стимулирует митоз, действие которых зависит от преобразование сигнала принимающей ткани или клеток. Другими словами, когда рецептор гормона на клетке-реципиенте стимулируется, сигнал передается от поверхности клетки к ядро клетки чтобы повлиять на некоторые изменения в регуляции транскрипции генов на ядерном уровне. Некоторые онкогены являются частью самой системы передачи сигнала или сигнального рецепторы в самих клетках и тканях, тем самым контролируя чувствительность к таким гормонам. Онкогены часто производят митогены, или участвуют в транскрипция ДНК в синтез белка, что создает белки и ферменты отвечает за производство продукции и биохимические вещества ячейки используют и взаимодействуют с.
Мутации в протоонкогенах, которые в норме являются пассивными аналогами онкогены, может изменить свои выражение и функция, увеличивая количество или активность белкового продукта. Когда это происходит, протоонкогены становятся онкогены, и этот переход нарушает нормальный баланс клеточный цикл регуляция в клетке, что делает возможным неконтролируемый рост. Невозможно снизить вероятность рака, удалив протоонкогены из геном, даже если бы это было возможно, поскольку они имеют решающее значение для роста, ремонта и гомеостаз организма. Только когда они мутируют, сигналы к росту становятся чрезмерными.
Один из первых онкогены быть определенным в исследования рака это онкоген ras. Мутации в семье Рас протоонкогены (включающие H-Ras, N-Ras и K-Ras) очень распространены и обнаруживаются в 20-30% всех опухолей человека.[87] Первоначально Ras был идентифицирован в геноме вируса саркомы Харви, и исследователи были удивлены тем, что этот ген не только присутствует в геноме человека, но и при лигировании со стимулирующим контрольным элементом он может вызывать рак в культурах клеточных линий.[88]
Протоонкогены
Протоонкогены способствуют росту клеток множеством способов. Многие могут производить гормоны, «химические посланники» между клетками, которые стимулируют митоз, эффект которых зависит от преобразование сигнала принимающей ткани или клеток. Некоторые отвечают за систему передачи сигнала и сигнал рецепторы в самих клетках и тканях, тем самым контролируя чувствительность к таким гормонам. Они часто производят митогены, или участвуют в транскрипция ДНК в синтез белка, которые создают белки и ферменты отвечает за производство продукции и биохимические вещества ячейки используют и взаимодействуют с.
Мутации в протоонкогенах могут изменять их выражение и функция, увеличивая количество или активность белкового продукта. Когда это происходит, они становятся онкогены, и, таким образом, клетки имеют более высокий шанс чрезмерного и неконтролируемого деления. Невозможно снизить вероятность рака, удалив протоонкогены из геном, так как они имеют решающее значение для роста, ремонта и гомеостаз тела. Только когда они мутируют, сигналы для роста становятся чрезмерными. Важно отметить, что ген, обладающий стимулирующей ролью роста, может увеличивать канцерогенный потенциал клетки при условии, что все необходимые клеточные механизмы, обеспечивающие рост, задействованы. активирован.[89] Это состояние также включает инактивацию определенных генов-супрессоров опухолей (см. Ниже). Если условие не выполняется, клетка может перестать расти и умирать. Это позволяет идентифицировать стадию и тип раковая клетка который растет под контролем данного онкогена, который имеет решающее значение для разработки стратегий лечения.
Гены-супрессоры опухолей
Гены-супрессоры опухолей код для сигналов антипролиферации и белков, подавляющих митоз и рост клеток. Обычно супрессоры опухолей факторы транскрипции которые активируются сотовыми стресс или повреждение ДНК. Часто повреждение ДНК вызывает присутствие свободно плавающего генетического материала, а также другие признаки и запускает ферменты и пути, которые приводят к активации гены-супрессоры опухолей. Функции таких генов заключаются в том, чтобы останавливать развитие клеточного цикла с целью восстановления ДНК, предотвращая передачу мутаций дочерним клеткам. В p53 белок, один из наиболее важных изученных генов-супрессоров опухолей, является фактором транскрипции, активируемым многими клеточными стрессорами, включая гипоксия и ультрафиолетовая радиация повреждение.
Несмотря на то, что почти половина всех видов рака, возможно, связана с изменениями в p53, его функция подавления опухолей плохо изучена. p53 явно выполняет две функции: одну ядерную роль как фактора транскрипции, а другую - цитоплазматическую роль в регуляции клеточного цикла, деления клеток и апоптоза.
В Гипотеза Варбурга является предпочтительным использованием гликолиза в качестве энергии для поддержания роста рака. Было показано, что р53 регулирует переход от дыхательного пути к гликолитическому.[90]
Однако мутация может повредить сам ген-супрессор опухоли или сигнальный путь, который его активирует, «выключив». Неизменным следствием этого является то, что восстановление ДНК затруднено или подавлено: повреждение ДНК накапливается без восстановления, что неизбежно приводит к раку.
Мутации генов-супрессоров опухолей, возникающие в зародышевый клетки передаются потомство, и увеличивают вероятность диагноза рака у последующих поколений. Члены этих семей имеют повышенную заболеваемость и уменьшение латентного периода множественных опухолей. Типы опухолей типичны для каждого типа мутации гена-супрессора опухоли, при этом одни мутации вызывают определенные виды рака, а другие мутации вызывают другие. Способ наследования мутантных опухолевых супрессоров заключается в том, что пораженный член наследует дефектную копию от одного родителя и нормальную копию от другого. Например, люди, унаследовавшие одного мутанта p53 аллель (и поэтому гетерозиготный для мутировавших p53) может развиваться меланомы и панкреатический рак, известный как Синдром Ли-Фраумени. Другие наследственные синдромы гена-супрессора опухоли включают: Руб. мутации, связанные с ретинобластома, и APC генные мутации, связанные с аденополипоз, рак толстой кишки. Аденополипозный рак толстой кишки связан с тысячами полипов толстой кишки в молодом возрасте, что приводит к рак толстой кишки в относительно раннем возрасте. Наконец, унаследованные мутации в BRCA1 и BRCA2 привести к раннему началу рак молочной железы.
В 1971 году было предложено, чтобы развитие рака зависело как минимум от двух мутационных событий. В том, что стало известно как Knudson гипотеза с двумя ударами, унаследованная мутация зародышевой линии в ген-супрессор опухоли вызовет рак только в том случае, если другое событие мутации произойдет позже в жизни организма, инактивируя другой аллель того, что ген-супрессор опухоли.[91]
Обычно онкогены доминирующий, поскольку они содержат мутации с усилением функции, а мутировавшие опухолевые супрессоры рецессивный, поскольку они содержат мутации с потерей функции. Каждая клетка имеет две копии одного и того же гена, по одной от каждого родителя, и в большинстве случаев усиления функциональных мутаций только в одной копии конкретного протоонкогена достаточно, чтобы сделать этот ген истинным онкогеном. С другой стороны, мутации потери функции должны произойти в обеих копиях гена-супрессора опухоли, чтобы сделать этот ген полностью нефункциональным. Однако бывают случаи, когда одна мутировавшая копия ген-супрессор опухоли может представить другой, дикого типа копировать нефункционально. Это явление называется доминирующий негативный эффект и наблюдается во многих мутациях р53.
Модель Кнудсона с двумя хитами недавно была оспорена несколькими исследователями. Инактивации одного аллеля некоторых генов-супрессоров опухолей достаточно, чтобы вызвать опухоль. Это явление называется гаплонедостаточность и был продемонстрирован рядом экспериментальных подходов. Опухоли, вызванные гаплонедостаточность обычно имеют более поздний возраст начала по сравнению с таковыми при двухстороннем процессе.[92]
Множественные мутации

В общем, для возникновения рака требуются мутации обоих типов генов. Например, мутация, ограниченная одним онкогеном, будет подавлена нормальным контролем митоза и генами-супрессорами опухоли. выдвинутый посредством Гипотеза Кнудсона.[3] Мутация только одного гена-супрессора опухоли также не вызовет рак из-за наличия многих "резервный "гены, которые дублируют его функции. Только когда достаточное количество протоонкогенов мутировало в онкогены, и достаточное количество генов-супрессоров опухолей деактивировано или повреждено, сигналы роста клеток подавляют сигналы, регулирующие его, и рост клеток быстро выходит из-под контроля. .[5] Часто, поскольку эти гены регулируют процессы, предотвращающие большинство повреждений самих генов, скорость мутаций увеличивается с возрастом, поскольку повреждение ДНК формирует Обратная связь петля.
Мутация генов-супрессоров опухолей, которые передаются следующему поколению не только клеток, но и их потомство, может повысить вероятность передачи рака по наследству. Члены этих семей имеют повышенную заболеваемость и снижение латентного периода множественных опухолей. Тип наследования мутантных супрессоров опухолей состоит в том, что пораженный член наследует дефектную копию от одного родителя и нормальную копию от другого. Поскольку мутации в супрессорах опухолей действуют рецессивным образом (обратите внимание, однако, есть исключения), потеря нормальной копии создает рак. фенотип. Например, люди, которые гетерозиготный мутации p53 часто становятся жертвами Синдром Ли-Фраумени, и которые гетерозиготны по Руб. мутации развиваются ретинобластома. Аналогичным образом мутации в аденоматозный полипоз кишечной палочки ген связаны с аденополипоз, рак толстой кишки, с тысячами полипов в толстой кишке в молодом возрасте, тогда как мутации в BRCA1 и BRCA2 привести к раннему началу рак молочной железы.
Новая идея, объявленная в 2011 году, - это крайняя версия множественных мутаций, названная хромотрипсис его сторонниками. Эта идея, затрагивающая только 2–3% случаев рака и до 25% случаев рака костей, включает в себя катастрофическое разрушение хромосомы на десятки или сотни частей, а затем ее неправильное исправление. Это разрушение, вероятно, происходит, когда хромосомы уплотняются во время нормальное деление клеток, но причина разрушения неизвестна. Согласно этой модели, рак возникает в результате одного изолированного события, а не в результате медленного накопления множества мутаций.[93]
Немутагенные канцерогены
Много мутагены являются также канцерогены, но некоторые канцерогены не являются мутагенами. Примеры канцерогенов, не являющихся мутагенами, включают: алкоголь и эстроген. Считается, что они способствуют развитию рака за счет своего стимулирующего воздействия на скорость клеточной митоз. Более высокие скорости митоза все больше оставляют меньше возможностей для репаративных ферментов для восстановления поврежденной ДНК во время Репликация ДНК, увеличивая вероятность генетической ошибки. Ошибка, допущенная во время митоза, может привести к тому, что дочерние клетки получат неправильное количество хромосомы, что приводит к анеуплоидия и может привести к раку.
Роль инфекций
Бактериальный
Helicobacter pylori может вызвать рак желудка. Хотя данные различаются по странам, в целом от 1% до 3% людей, инфицированных Helicobacter pylori заболевают раком желудка в течение жизни по сравнению с 0,13% людей, которые не имели Хеликобактер пилори инфекция.[94][95] Хеликобактер пилори инфекция очень распространена. По оценке 2002 года, он присутствует в тканях желудка 74% взрослых людей среднего возраста в развивающихся странах и 58% в развитых странах.[96] Поскольку от 1% до 3% инфицированных людей могут заболеть раком желудка,[97] Хеликобактер пилори-индуцированный рак желудка является третьей по величине причиной смертности от рака в мире по состоянию на 2018 год.[98]
Заражение Хеликобактер пилори не вызывает никаких симптомов примерно у 80% инфицированных.[99] Около 75% людей инфицированы Хеликобактер пилори развиваться гастрит.[100] Таким образом, обычное следствие Хеликобактер пилори Инфекция - хронический бессимптомный гастрит.[101] Из-за обычного отсутствия симптомов, когда наконец диагностируется рак желудка, он часто оказывается довольно запущенным. Более чем у половины пациентов с раком желудка на момент постановки диагноза обнаруживаются метастазы в лимфатические узлы.[102]
Гастрит, вызванный Хеликобактер пилори сопровождается воспаление, характеризующийся проникновением нейтрофилы и макрофаги в эпителий желудка, что способствует накоплению провоспалительные цитокины и активные формы кислорода /активные формы азота (ROS / RNS).[103] Существенное присутствие ROS / RNS вызывает повреждение ДНК, включая 8-оксо-2'-дезоксигуанозин (8-OHdG).[103] Если заражение Хеликобактер пилори нести цитотоксический cagA ген (присутствует примерно в 60% западных изолятов и более высоком проценте азиатских изолятов), они могут повышать уровень 8-OHdG в клетках желудка в 8 раз, в то время как если Хеликобактер пилори не несут ген cagA, увеличение 8-OHdG примерно в 4 раза.[104] В добавок к окислительное повреждение ДНК 8-OHdG, Хеликобактер пилори инфекция вызывает другие характерные повреждения ДНК, включая двухцепочечные разрывы ДНК.[105]
Хеликобактер пилори также вызывает многие эпигенетический изменения, связанные с развитием рака.[106][107] Эти эпигенетический изменения связаны с Хеликобактер пилори-индуцированный метилирование CpG-сайтов в промоторах генов[106] и Хеликобактер пилори-индуцированное измененное выражение нескольких микроРНК.[107]
В обзоре Сантоса и Рибейро[108] Хеликобактер пилори инфекция связана с эпигенетически сниженной эффективностью механизма репарации ДНК, что способствует накоплению мутаций и геномной нестабильности, а также желудочному канцерогенезу. В частности, Raza et al.[109] показали, что экспрессия двух белков репарации ДНК, ERCC1 и PMS2, был однажды сильно сокращен Хеликобактер пилори инфекция прогрессировала, чтобы вызвать диспепсия. Диспепсия встречается примерно у 20% инфицированных людей.[110] Кроме того, согласно обзору Raza et al.,[109] инфекция желудка человека с Хеликобактер пилори вызывает эпигенетически сниженную экспрессию белков репарации ДНК MLH1, MGMT и MRE11. Снижение репарации ДНК в присутствии повышенного повреждения ДНК увеличивает канцерогенные мутации и, вероятно, является важной причиной Хеликобактер пилори канцерогенез.
Вирусный
Кроме того, многие виды рака возникают из-за вирусный инфекция; это особенно верно для таких животных, как птицы, но в меньшей степени люди. 12% случаев рака у человека можно отнести к вирусной инфекции.[111] Типы вирусно-индуцированных опухолей можно разделить на два: резко преобразующий или медленно трансформируется. В остро трансформирующихся вирусах вирусные частицы несут ген, который кодирует сверхактивный онкоген, называемый вирусным онкогеном (v-onc), и инфицированная клетка трансформируется, как только v-onc экспрессируется. Напротив, в медленно трансформирующихся вирусах геном вируса вставляется, тем более что вставка вирусного генома является обязательной частью ретровирусы, рядом с протоонкогеном в геноме хозяина. Вирусный промоутер или другие элементы регуляции транскрипции, в свою очередь, вызывают сверхэкспрессию этого протоонкогена, что, в свою очередь, вызывает неконтролируемую клеточную пролиферацию. Поскольку вставка вирусного генома не специфична для протоонкогенов и вероятность вставки рядом с этим протоонкогеном мала, медленно трансформирующиеся вирусы имеют очень долгую латентность опухоли по сравнению с быстро трансформирующимся вирусом, который уже несет вирусный онкоген.
Вирусы, которые, как известно, вызывают рак, такие как ВПЧ (рак шейки матки ), Гепатит Б (рак печени ), и EBV (тип лимфома ), все ДНК-вирусы. Считается, что когда вирус заражает клетку, он вставляет часть своей собственной ДНК рядом с генами роста клетки, вызывая деление клетки. Группа измененных клеток, которые образуются в результате первого деления клеток, имеют одинаковую вирусную ДНК рядом с генами роста клеток. Группа измененных клеток теперь особенная, потому что один из нормальных средств контроля роста был утерян.
В зависимости от своего местоположения клетки могут быть повреждены радиацией, химическими веществами из сигаретного дыма и воспалениями в результате бактериальной инфекции или других вирусов. Каждая ячейка имеет шанс повреждения. Клетки часто умирают, если они повреждены, из-за сбоя жизненно важных процессов или иммунной системы, однако иногда повреждение приводит к нокауту одного гена рака. У старого человека есть тысячи, десятки тысяч или сотни тысяч выбитых клеток. Вероятность того, что у кого-то возникнет рак, очень мала.[нужна цитата ]
Когда повреждение происходит в любой области измененных клеток, происходит нечто иное. Каждая из клеток имеет потенциал роста. Измененные клетки будут делиться быстрее, если область повреждена физическими, химическими или вирусными агентами. А порочный круг был настроен: повреждение области приведет к разделению измененных клеток, что повысит вероятность того, что они будут нокаутированы.
Эта модель канцерогенеза популярна, потому что она объясняет, почему рак растет. Можно было бы ожидать, что клетки, поврежденные радиацией, умрут или, по крайней мере, будут жить хуже, потому что у них меньше работающих генов; вирусы увеличивают количество работающих генов.
Одна мысль состоит в том, что в конечном итоге у нас могут появиться тысячи вакцин, чтобы предотвратить каждый вирус, который может изменить наши клетки. Вирусы могут по-разному влиять на разные части тела. Возможно, удастся предотвратить множество различных видов рака, иммунизируя против одного вирусного агента. Например, вполне вероятно, что ВПЧ играет роль в развитии рака слизистой оболочки рта.
Гельминтоз
Известно, что некоторые паразитические черви обладают канцерогенными свойствами.[112] Они включают:
- Clonorchis sinensis (организм, вызывающий Клонорхоз ) и Описторхис виверрини (вызывая Описторхоз ) связаны с холангиокарцинома.[113]
- Шистосома виды (организмы, вызывающие Шистосомоз ) связан с Рак мочевого пузыря.
Эпигенетика
Эпигенетика - это изучение регуляции экспрессии генов посредством химических немутационных изменений в структуре ДНК. Теория эпигенетика в патогенезе рака заключается в том, что немутационные изменения ДНК могут приводить к изменениям в экспрессии генов. Как обычно, онкогены молчат, например, из-за Метилирование ДНК. Потеря этого метилирования может вызвать аберрантную экспрессию онкогены, приводящие к патогенезу рака. Известные механизмы эпигенетических изменений включают: Метилирование ДНК, и метилирование или ацетилирование гистон белки, связанные с хромосомной ДНК в определенных местах. Классы лекарств, известные как Ингибиторы HDAC и ДНК-метилтрансфераза ингибиторы, могут повторно регулировать эпигенетическую передачу сигналов в раковая клетка.
Эпимутации включают метилирование или деметилирование Острова CpG из промоутер области генов, которые приводят к репрессии или дерепрессии, соответственно, экспрессии генов.[114][115][116] Эпимутации также могут происходить путем ацетилирования, метилирования, фосфорилирования или других изменений гистонов, создавая гистоновый код который подавляет или активирует экспрессию генов, и такие эпимутации гистонов могут быть важными эпигенетическими факторами при раке.[117][118] Кроме того, канцерогенная эпимутация может происходить из-за изменений архитектуры хромосом, вызванных такими белками, как HMGA2.[119] Еще одним источником эпимутации является повышенная или пониженная экспрессия микроРНК (миРНК). Например, дополнительная экспрессия miR-137 может вызывать подавление экспрессии 491 гена, а miR-137 эпигенетически подавляется в 32% случаев колоректального рака>[8]
Раковые стволовые клетки
Новый взгляд на канцерогенез основан на интеграции идей биология развития в онкология. В раковые стволовые клетки гипотеза предлагает, чтобы различные виды ячеек в неоднородный опухоль возникает из одной клетки, называемой раковой стволовой клеткой. Раковые стволовые клетки могут возникнуть в результате трансформации взрослые стволовые клетки или дифференцированный клетки в теле. Эти клетки сохраняются как субкомпонент опухоли и сохраняют ключевые свойства стволовых клеток. Они дают начало множеству клеток, способны к самообновлению и гомеостатический контроль.[120] Кроме того, рецидив рака и появления метастаз также относятся к этим клеткам. В раковые стволовые клетки гипотеза не противоречит более ранним представлениям о канцерогенезе. Гипотеза раковых стволовых клеток была предложенным механизмом, который способствует неоднородность опухоли.
Клональная эволюция
Хотя генетические и эпигенетический изменения в генах-супрессорах опухолей и онкогенах изменяют поведение клеток, эти изменения, в конце концов, приводят к раку из-за их воздействия на популяцию неопластический клетки и их микросреда.[58] Мутантные клетки в новообразованиях конкурируют за пространство и ресурсы. Таким образом, клон с мутацией в гене-супрессоре опухоли или онкогене будет размножаться только в новообразовании, если эта мутация дает клону конкурентное преимущество перед другими клонами и нормальными клетками в его микроокружении.[121] Таким образом, процесс канцерогенеза формально является дарвиновским процессом. эволюция, известный как соматическая или клональная эволюция.[59] Кроме того, в свете дарвинистских механизмов канцерогенеза было высказано предположение, что различные формы рака можно разделить на пубертатные и геронтологические. В настоящее время проводятся антропологические исследования рака как естественного эволюционного процесса, посредством которого естественный отбор уничтожает экологически неполноценные фенотипы, поддерживая другие. Согласно этой теории, рак бывает двух разных типов: от рождения до конца полового созревания (примерно 20 лет), телеологически склонный к поддерживающей групповой динамике, и от среднего возраста до смерти (примерно возраст 40+), телеологически склонный к удалению от перенаселенной группы. динамика.[нужна цитата ]
Смотрите также
использованная литература
- ^ Томасетти К., Ли Л., Фогельштейн Б. (23 марта 2017 г.). «Деление стволовых клеток, соматические мутации, этиология рака и профилактика рака». Наука. 355 (6331): 1330–1334. Bibcode:2017Научный ... 355.1330Т. Дои:10.1126 / science.aaf9011. ЧВК 5852673. PMID 28336671.
- ^ а б Вуд Л.Д., Парсонс Д.В., Джонс С., Лин Дж., Сьоблом Т., Лири Р.Дж. и др. (Ноябрь 2007 г.). «Геномные пейзажи человеческого рака груди и колоректального рака». Наука. 318 (5853): 1108–13. Bibcode:2007Sci ... 318.1108W. CiteSeerX 10.1.1.218.5477. Дои:10.1126 / science.1145720. PMID 17932254.
- ^ а б Knudson AG (ноябрь 2001 г.). «Два генетических хита (более или менее) до рака». Обзоры природы. Рак. 1 (2): 157–62. Дои:10.1038/35101031. PMID 11905807.
- ^ Фирон ER, Фогельштейн Б. (июнь 1990 г.). «Генетическая модель колоректального туморогенеза». Ячейка. 61 (5): 759–67. Дои:10.1016 / 0092-8674 (90) 90186-И. PMID 2188735.
- ^ а б c Беликов, Алексей В. (22 сентября 2017 г.). «Количество ключевых канцерогенных событий можно предсказать по заболеваемости раком». Научные отчеты. 7 (1): 12170. Bibcode:2017НатСР ... 712170Б. Дои:10.1038 / s41598-017-12448-7. ЧВК 5610194. PMID 28939880.
- ^ Croce CM (январь 2008 г.). «Онкогены и рак». Медицинский журнал Новой Англии. 358 (5): 502–11. Дои:10.1056 / NEJMra072367. PMID 18234754.
- ^ Лим LP, Лау NC, Гарретт-Энгеле П., Гримсон А., Шелтер Дж. М., Касл Дж., Бартель Д. П., Линсли П. С., Джонсон Дж. М. (февраль 2005 г.). «Анализ микроматрицы показывает, что некоторые микроРНК подавляют большое количество целевых мРНК». Природа. 433 (7027): 769–73. Bibcode:2005Натура.433..769L. Дои:10.1038 / природа03315. PMID 15685193.
- ^ а б Балагер Ф., Ссылка А, Лозано Дж. Дж., Куатрекасас М., Нагасака Т., Боланд Ч. Р., Гоэль А (август 2010 г.). «Эпигенетическое подавление miR-137 является ранним событием в колоректальном канцерогенезе». Исследования рака. 70 (16): 6609–18. Дои:10.1158 / 0008-5472.CAN-10-0622. ЧВК 2922409. PMID 20682795.
- ^ Кастан МБ (апрель 2008 г.). «Реакция на повреждение ДНК: механизмы и роль в человеческих заболеваниях: лекция 2007 г. на присуждении премии имени Г.А. Клоуса». Молекулярные исследования рака. 6 (4): 517–24. Дои:10.1158 / 1541-7786.MCR-08-0020. PMID 18403632.
- ^ а б Каннингем Ф. Х., Фибелкорн С., Джонсон М., Мередит С. (ноябрь 2011 г.). «Новое применение подхода маржи воздействия: разделение токсичных веществ табачного дыма». Пищевая и химическая токсикология. 49 (11): 2921–33. Дои:10.1016 / j.fct.2011.07.019. PMID 21802474.
- ^ Канави HE, Герстенблит MR (декабрь 2011 г.). «Ультрафиолетовое излучение и меланома». Семинары по кожной медицине и хирургии. 30 (4): 222–8. Дои:10.1016 / j.sder.2011.08.003. PMID 22123420.
- ^ Ханда О, Наито Й, Йошикава Т. (2011). «Редокс-биология и канцерогенез желудка: роль Helicobacter pylori». Редокс-отчет. 16 (1): 1–7. Дои:10,1179 / 174329211X12968219310756. PMID 21605492.
- ^ Смела М.Э., Хамм М.Л., Хендерсон П.Т., Харрис С.М., Харрис TM, Эссигманн Дж. М. (май 2002 г.). «Аддукт афлатоксина B (1) с формамидопиримидином играет важную роль в возникновении типов мутаций, наблюдаемых при гепатоцеллюлярной карциноме человека». Труды Национальной академии наук Соединенных Штатов Америки. 99 (10): 6655–60. Bibcode:2002PNAS ... 99.6655S. Дои:10.1073 / pnas.102167699. ЧВК 124458. PMID 12011430.
- ^ Кацурано М., Нива Т., Ясуи Ю., Сигемацу Ю., Ямасита С., Такешима Х., Ли М.С., Ким Ю.Дж., Танака Т., Ушидзима Т. (январь 2012 г.). «Ранняя стадия формирования дефекта эпигенетического поля в модели колита у мышей и несущественные роли Т- и В-клеток в индукции метилирования ДНК». Онкоген. 31 (3): 342–51. Дои:10.1038 / onc.2011.241. PMID 21685942.
- ^ Бернштейн С., Голубек Х., Бхаттачарья А.К., Нгуен Х., Пейн С.М., Зейтлин Б., Бернштейн Х. (август 2011 г.). «Канцерогенность дезоксихолата, вторичной желчной кислоты». Архив токсикологии. 85 (8): 863–71. Дои:10.1007 / s00204-011-0648-7. ЧВК 3149672. PMID 21267546.
- ^ Малкин Д (апрель 2011). «Синдром Ли-Фраумени». Гены и рак. 2 (4): 475–84. Дои:10.1177/1947601911413466. ЧВК 3135649. PMID 21779515.
- ^ Fearon ER (ноябрь 1997 г.). «Синдромы рака человека: ключи к разгадке происхождения и природы рака». Наука. 278 (5340): 1043–50. Bibcode:1997Sci ... 278.1043F. Дои:10.1126 / science.278.5340.1043. PMID 9353177.
- ^ Лихтенштейн П., Холм Н.В., Веркасало П.К., Илиадоу А., Каприо Дж., Коскенвуо М., Пуккала Е., Скайтте А., Хемминки К. (июль 2000 г.). «Экологические и наследственные факторы в возникновении рака - анализ когорт близнецов из Швеции, Дании и Финляндии». Медицинский журнал Новой Англии. 343 (2): 78–85. Дои:10.1056 / NEJM200007133430201. PMID 10891514.
- ^ Halford S, Rowan A, Sawyer E, Talbot I, Tomlinson I (июнь 2005 г.). «O (6) -метилгуанинметилтрансфераза при колоректальном раке: обнаружение мутаций, потеря экспрессии и слабая связь с переходами G: C> A: T». Кишечник. 54 (6): 797–802. Дои:10.1136 / гут.2004.059535. ЧВК 1774551. PMID 15888787.
- ^ а б Нараянан Л., Фритцелл Дж. А., Бейкер С. М., Лискай Р. М., Глейзер П. М. (апрель 1997 г.). «Повышенные уровни мутаций во многих тканях мышей с дефицитом гена репарации несоответствия ДНК Pms2». Труды Национальной академии наук Соединенных Штатов Америки. 94 (7): 3122–7. Bibcode:1997ПНАС ... 94.3122Н. Дои:10.1073 / пнас.94.7.3122. ЧВК 20332. PMID 9096356.
- ^ а б Хеган Д.К., Нараянан Л., Джирик FR, Эдельманн В., Лискай Р.М., Глейзер П.М. (декабрь 2006 г.). «Различия в паттернах генетической нестабильности у мышей, лишенных генов репарации несовпадений Pms2, Mlh1, Msh2, Msh3 и Msh6». Канцерогенез. 27 (12): 2402–8. Дои:10.1093 / carcin / bgl079. ЧВК 2612936. PMID 16728433.
- ^ а б Тутт А.Н., ван Остром К.Т., Росс Г.М., ван Стиг Х., Эшворт А. (март 2002 г.). «Нарушение Brca2 увеличивает скорость спонтанных мутаций in vivo: синергизм с ионизирующим излучением». EMBO отчеты. 3 (3): 255–60. Дои:10.1093 / embo-reports / kvf037. ЧВК 1084010. PMID 11850397.
- ^ Герман Дж. (Март 1969 г.). «Синдром Блума. I. Генетические и клинические наблюдения у первых двадцати семи пациентов». Американский журнал генетики человека. 21 (2): 196–227. ЧВК 1706430. PMID 5770175.
- ^ О'Хаган Х.М., Мохаммад Х.П., Бейлин С.Б. (август 2008 г.). Ли JT (ред.). «Двухцепочечные разрывы могут инициировать сайленсинг генов и SIRT1-зависимое начало метилирования ДНК в экзогенном промоторном острове CpG». PLOS Genetics. 4 (8): e1000155. Дои:10.1371 / journal.pgen.1000155. ЧВК 2491723. PMID 18704159.
- ^ Cuozzo C, Porcellini A, Angrisano T, Morano A, Lee B, Di Pardo A, Messina S, Iuliano R, Fusco A, Santillo MR, Muller MT, Chiariotti L, Gottesman ME, Avvedimento EV (июль 2007 г.). «Повреждение ДНК, гомологически направленная репарация и метилирование ДНК». PLOS Genetics. 3 (7): e110. Дои:10.1371 / journal.pgen.0030110. ЧВК 1913100. PMID 17616978.
- ^ Вильнев П.Дж., Мао Ю. (ноябрь 1994 г.). «Пожизненная вероятность развития рака легких по курению, Канада». Канадский журнал общественного здравоохранения. 85 (6): 385–8. PMID 7895211.
- ^ Герлингер М., Роуэн А.Дж., Хорсвелл С., Ларкин Дж., Эндесфельдер Д., Гронроос Э. и др. (Март 2012 г.). «Внутриопухолевая гетерогенность и разветвленная эволюция, выявленные с помощью мультирегионального секвенирования». Медицинский журнал Новой Англии. 366 (10): 883–92. Дои:10.1056 / NEJMoa1113205. ЧВК 4878653. PMID 22397650.
- ^ а б Лопес-Ласаро М. (август 2015 г.). "Теория деления стволовых клеток рака". Клеточный цикл. 14 (16): 2547–8. Дои:10.1080/15384101.2015.1062330. ЧВК 5242319. PMID 26090957.
- ^ а б c Лопес-Ласаро М (май 2015 г.). «Способность стволовых клеток к миграции может объяснить существование рака неизвестной первичной локализации. Переосмысление метастазов». Онкология. 2 (5): 467–75. Дои:10.18632 / oncoscience.159. ЧВК 4468332. PMID 26097879.
- ^ Томасетти С., Фогельштейн Б. (январь 2015 г.). «Этиология рака. Различия в риске рака среди тканей можно объяснить количеством делений стволовых клеток». Наука. 347 (6217): 78–81. Дои:10.1126 / science.1260825. ЧВК 4446723. PMID 25554788.
- ^ Slaughter DP, Southwick HW, Smejkal W (сентябрь 1953 г.). «Полевая канцеризация в многослойном плоском эпителии полости рта; клинические последствия мультицентрического происхождения». Рак. 6 (5): 963–8. Дои:10.1002 / 1097-0142 (195309) 6: 5 <963 :: AID-CNCR2820060515> 3.0.CO; 2-Q. PMID 13094644.
- ^ Бернштейн С., Бернштейн Х., Пейн С.М., Дворжак К., Гарвал Х. (февраль 2008 г.). «Дефекты поля в прогрессировании рака желудочно-кишечного тракта». обзор. Письма о раке. 260 (1–2): 1–10. Дои:10.1016 / j.canlet.2007.11.027. ЧВК 2744582. PMID 18164807.
- ^ Нгуен Х., Лустаунау С., Фасиста А., Рэмси Л., Хассуна Н., Тейлор Х., Кроуз Р., Пейн С.М., Цикитис В.Л., Гольдшмид С., Банерджи Б., Перини Р.Ф., Бернштейн С. (2010). «Дефицит Pms2, ERCC1, Ku86, CcOI в полевых дефектах при прогрессировании рака толстой кишки». Журнал визуализированных экспериментов (41): 1931. Дои:10.3791/1931. ЧВК 3149991. PMID 20689513.
- ^ Рубин Х (март 2011 г.). «Поля и полевая канцеризация: пренеопластическое происхождение рака: бессимптомные гиперпластические поля являются предшественниками неоплазии, и их прогрессирование в опухоли можно отслеживать по плотности насыщения в культуре». BioEssays. 33 (3): 224–31. Дои:10.1002 / bies.201000067. PMID 21254148.
- ^ Цао Дж. Л., Ятабэ Ю., Саловаара Р., Ярвинен Х. Дж., Меклин Дж. П., Аалтонен Л. А., Таваре С., Шибата Д. (февраль 2000 г.). «Генетическая реконструкция истории индивидуальных колоректальных опухолей». Труды Национальной академии наук Соединенных Штатов Америки. 97 (3): 1236–41. Bibcode:2000PNAS ... 97.1236T. Дои:10.1073 / pnas.97.3.1236. ЧВК 15581. PMID 10655514.
- ^ а б c Фогельштейн Б., Пападопулос Н., Велкулеску В.Э., Чжоу С., Диас Л.А., Кинзлер К.В. (март 2013 г.). «Пейзажи генома рака». обзор. Наука. 339 (6127): 1546–58. Bibcode:2013Научный ... 339.1546V. Дои:10.1126 / наука.1235122. ЧВК 3749880. PMID 23539594.
- ^ Шен Л., Кондо Й., Роснер Г.Л., Сяо Л., Эрнандес Н.С., Вилайтонг Дж., Хулихан П.С., Кроуз Р.С., Прасад А.Р., Эйнспар Дж. Г., Бакмайер Дж., Альбертс Д.С., Гамильтон С.Р., Исса Дж. П. (сентябрь 2005 г.). «Метилирование промотора MGMT и дефект поля при спорадическом колоректальном раке». Журнал Национального института рака. 97 (18): 1330–8. Дои:10.1093 / jnci / dji275. PMID 16174854.
- ^ а б Ли К. Х., Ли Дж. С., Нам Дж. Х., Чхве С., Ли МС, Пак С. С., Джунг С. В., Ли Дж. Х. (октябрь 2011 г.). «Статус метилирования промотора генов hMLH1, hMSH2 и MGMT при колоректальном раке, ассоциированном с последовательностью аденома-карцинома». Архив хирургии Лангенбека. 396 (7): 1017–26. Дои:10.1007 / s00423-011-0812-9. PMID 21706233.
- ^ Сврчек М., Бухард О., Колас С., Куле Ф., Дюмон С., Массауди И. и др. (Ноябрь 2010 г.). «Толерантность к метилированию из-за дефекта поля O6-метилгуанин-ДНК-метилтрансферазы (MGMT) в слизистой оболочке толстой кишки: начальный шаг в развитии колоректального рака с дефицитом репарации несоответствия». Кишечник. 59 (11): 1516–26. Дои:10.1136 / gut.2009.194787. PMID 20947886.
- ^ а б c d Фасиста А., Нгуен Х., Льюис С., Прасад А.Р., Рэмси Л., Зейтлин Б., Нфонсам В., Кроуз Р.С., Бернштейн Х., Пейн С.М., Стерн С., Оатман Н., Банерджи Б., Бернштейн С. (апрель 2012 г.). «Недостаточная экспрессия ферментов репарации ДНК при раннем прогрессировании до спорадического рака толстой кишки». Целостность генома. 3 (1): 3. Дои:10.1186/2041-9414-3-3. ЧВК 3351028. PMID 22494821.
- ^ Paluszczak J, Misiak P, Wierzbicka M, Woniak A, Baer-Dubowska W (февраль 2011 г.). «Частое гиперметилирование DAPK, RARbeta, MGMT, RASSF1A и FHIT при плоскоклеточном раке гортани и прилегающей нормальной слизистой оболочке». Оральная онкология. 47 (2): 104–7. Дои:10.1016 / j.oraloncology.2010.11.006. PMID 21147548.
- ^ Zuo C, Zhang H, Spencer HJ, Vural E, Suen JY, Schichman SA, Smoller BR, Kokoska MS, Fan CY (октябрь 2009 г.). «Повышенная микросателлитная нестабильность и эпигенетическая инактивация гена hMLH1 при плоскоклеточной карциноме головы и шеи». Отоларингология - хирургия головы и шеи. 141 (4): 484–90. Дои:10.1016 / j.otohns.2009.07.007. PMID 19786217.
- ^ Тауфик Х.М., Эль-Максуд Н.М., Хак Б.Х., Эль-Щербины Ю.М. (2011). «Плоскоклеточный рак головы и шеи: иммуногистохимия восстановления несоответствия и гиперметилирование промотора гена hMLH1». Американский журнал отоларингологии. 32 (6): 528–36. Дои:10.1016 / j.amjoto.2010.11.005. PMID 21353335.
- ^ Zou XP, Zhang B, Zhang XQ, Chen M, Cao J, Liu WJ (ноябрь 2009 г.). «Промотор гиперметилирования нескольких генов в ранней аденокарциноме желудка и предраковых поражениях». Патология человека. 40 (11): 1534–42. Дои:10.1016 / j.humpath.2009.01.029. PMID 19695681.
- ^ Вани М., Афрозе Д., Махдуми М., Хамид И., Вани Б., Бхат Г., Вани Р., Вани К. (2012). «Статус метилирования промотора гена репарации ДНК (hMLH1) у пациентов с карциномой желудка в Кашмирской долине» (PDF). Азиатско-Тихоокеанский журнал профилактики рака. 13 (8): 4177–81. Дои:10.7314 / APJCP.2012.13.8.4177. PMID 23098428.
- ^ Агарвал А., Полинени Р., Хусейн З., Вигода И., Бхагат Т.Д., Бхаттачарья С., Майтра А., Верма А. (2012). «Роль эпигенетических изменений в патогенезе пищевода Барретта и аденокарциномы пищевода». Международный журнал клинической и экспериментальной патологии. 5 (5): 382–96. ЧВК 3396065. PMID 22808291. Обзор.
- ^ Hofstad B, Vatn MH, Andersen SN, Huitfeldt HS, Rognum T., Larsen S, Osnes M (сентябрь 1996 г.). «Рост колоректальных полипов: повторное обнаружение и оценка нерезецированных полипов в течение трех лет». Кишечник. 39 (3): 449–56. Дои:10.1136 / гут.39.3.449. ЧВК 1383355. PMID 8949653.
- ^ Шмитт MW, Prindle MJ, Loeb LA (сентябрь 2012 г.). «Последствия генетической гетерогенности рака». Летопись Нью-Йоркской академии наук. 1267 (1): 110–6. Bibcode:2012НЯСА1267..110С. Дои:10.1111 / j.1749-6632.2012.06590.x. ЧВК 3674777. PMID 22954224.
- ^ Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J и др. (Февраль 2001 г.). "Начальная последовательность и анализ человеческого генома". Природа. 409 (6822): 860–921. Bibcode:2001Натура.409..860л. Дои:10.1038/35057062. PMID 11237011.
- ^ Йост С.Е., Смит Э. Н., Шваб Р. Б., Бао Л., Юнг Х., Ван X, Восток Э., Пирс Дж. П., Мессер К., Паркер Б. А., Харисменди О., Фрейзер К. А. (август 2012 г.). «Идентификация соматических мутаций с высокой степенью достоверности во всей последовательности генома фиксированных формалином образцов рака молочной железы». Исследования нуклеиновых кислот. 40 (14): e107. Дои:10.1093 / нар / gks299. ЧВК 3413110. PMID 22492626.
- ^ Бергер М.Ф., Ходис Э., Хеффернан Т.П., Дерибе Ю.Л., Лоуренс М.С., Протопопов А. и др. (Май 2012 г.). «Секвенирование генома меланомы выявляет частые мутации PREX2». Природа. 485 (7399): 502–6. Bibcode:2012Натура.485..502Б. Дои:10.1038 / природа11071. ЧВК 3367798. PMID 22622578.
- ^ Расник Д., Дюсберг PH (июнь 1999 г.). «Как анеуплоидия влияет на метаболический контроль и вызывает рак». Биохимический журнал. 340 (3): 621–30. Дои:10.1042/0264-6021:3400621. ЧВК 1220292. PMID 10359645.
- ^ а б c Лопес-Ласаро М. (март 2010 г.). «Новый взгляд на канцерогенез и альтернативный подход к терапии рака». Молекулярная медицина. 16 (3–4): 144–53. Дои:10.2119 / молмед.2009.00162. ЧВК 2802554. PMID 20062820.
- ^ Сото AM, Sonnenschein C (октябрь 2004 г.). «Теория соматических мутаций рака: растущие проблемы с парадигмой?». BioEssays. 26 (10): 1097–107. Дои:10.1002 / bies.20087. PMID 15382143.
- ^ Davies PC, Lineweaver CH (февраль 2011 г.). «Раковые опухоли как у Metazoa 1.0: выявление генов древних предков». Физическая биология. 8 (1): 015001. Bibcode:2011ФБио ... 8а5001Д. Дои:10.1088/1478-3975/8/1/015001. ЧВК 3148211. PMID 21301065.
- ^ Дин, Тим. «Рак похож на жизнь 1 миллиард лет назад, - говорят астробиологи», Австралийский ученый-биолог, 8 февраля 2011 г. Проверено 15 февраля 2011 г.
- ^ Стеррер, Вт (август 2016 г.). "Рак - мутационное воскрешение эндокариотических остатков" (PDF). Гипотезы рака. 1 (1): 1–15.
- ^ а б Nowell PC (октябрь 1976 г.). «Клональная эволюция популяций опухолевых клеток». Наука. 194 (4260): 23–8. Bibcode:1976Научный ... 194 ... 23N. Дои:10.1126 / science.959840. PMID 959840.
- ^ а б Мерло Л. М., Пеппер Дж. В., Рид Б. Дж., Мали СС (декабрь 2006 г.). «Рак как эволюционно-экологический процесс». Обзоры природы. Рак. 6 (12): 924–35. Дои:10.1038 / nrc2013. PMID 17109012.
- ^ Ханахан Д., Вайнберг Р.А. (январь 2000 г.). «Признаки рака». Ячейка. 100 (1): 57–70. Дои:10.1016 / S0092-8674 (00) 81683-9. PMID 10647931.
- ^ Чо РУ, Кларк М.Ф. (февраль 2008 г.). «Последние достижения в области стволовых раковых клеток». Текущее мнение в области генетики и развития. 18 (1): 48–53. Дои:10.1016 / j.gde.2008.01.017. PMID 18356041.
- ^ Танигучи К., Ву Л.В., Гривенников С.И., де Йонг П.Р., Лиан И., Юй FX, Ван К., Хо С.Б., Боланд Б.С., Чанг Дж.Т., Сандборн В.Дж., Хардиман Дж., Раз Э, Маэхара Ю., Йошимура А., Цукман-Росси Дж. , Гуань К.Л., Карин М. (март 2015 г.). «Модуль gp130-Src-YAP связывает воспаление с регенерацией эпителия». Природа. 519 (7541): 57–62. Bibcode:2015Натура.519 ... 57т. Дои:10.1038 / природа14228. ЧВК 4447318. PMID 25731159.
- ^ Ю Х, Лей П., Андредис С. Т. (декабрь 2013 г.). «JNK - новый регулятор межклеточной адгезии». Тканевые барьеры. 1 (5): e26845. Дои:10.4161 / tisb.26845. ЧВК 3942331. PMID 24868495.
- ^ Бусилло Дж. М., Аззам К. М., Цидловски Дж. А. (ноябрь 2011 г.). «Глюкокортикоиды повышают чувствительность врожденной иммунной системы за счет регуляции инфламмасомы NLRP3». Журнал биологической химии. 286 (44): 38703–13. Дои:10.1074 / jbc.M111.275370. ЧВК 3207479. PMID 21940629.
- ^ Ван Y, Bugatti M, Ulland TK, Vermi W, Gilfillan S, Colonna M (март 2016 г.). «Неизбыточная роль производного кератиноцитов IL-34 и нейтрофильного CSF1 в обновлении клеток Лангерганса в стабильном состоянии и во время воспаления». Европейский журнал иммунологии. 46 (3): 552–9. Дои:10.1002 / eji.201545917. ЧВК 5658206. PMID 26634935.
- ^ Сикейра Миетто Б., Кронер А., Джиролами Е.И., Сантос-Ногейра Е., Чжан Дж., Дэвид С. (декабрь 2015 г.). «Роль IL-10 в разрешении воспаления и функциональном восстановлении после повреждения периферических нервов». Журнал неврологии. 35 (50): 16431–42. Дои:10.1523 / JNEUROSCI.2119-15.2015. ЧВК 6605511. PMID 26674868.
- ^ Зайферт А.В., Маден М (2014). «Новые взгляды на регенерацию кожи позвоночных». Международный обзор клеточной и молекулярной биологии. 310. С. 129–69. Дои:10.1016 / B978-0-12-800180-6.00004-9. ISBN 978-0-12-800180-6. PMID 24725426.
- ^ Kwon MJ, Shin HY, Cui Y, Kim H, Thi AH, Choi JY, Kim EY, Hwang DH, Kim BG (декабрь 2015 г.). «CCL2 опосредует взаимодействия нейронов и макрофагов для активации прорегенеративных макрофагов после прекондиционирующей травмы». Журнал неврологии. 35 (48): 15934–47. Дои:10.1523 / JNEUROSCI.1924-15.2015. ЧВК 6605453. PMID 26631474.
- ^ Хаджишенгаллис Г., Чавакис Т. (январь 2013 г.). «Эндогенные модуляторы рекрутирования воспалительных клеток». Тенденции в иммунологии. 34 (1): 1–6. Дои:10.1016 / j.it.2012.08.003. ЧВК 3703146. PMID 22951309.
- ^ Нельсон А.М., Кацефф А.С., Рэтлифф Т.С., Гарза Л.А. (февраль 2016 г.). «Интерлейкин 6 и STAT3 регулируют экспрессию изоформы p63 в кератиноцитах во время регенерации». Экспериментальная дерматология. 25 (2): 155–7. Дои:10.1111 / exd.12896. ЧВК 4724264. PMID 26566817.
- ^ Видаль П.М., Лемменс Э., Дули Д., Хендрикс С. (февраль 2013 г.). «Роль« противовоспалительных »цитокинов в регенерации аксонов». Отзывы о цитокинах и факторах роста. 24 (1): 1–12. Дои:10.1016 / j.cytogfr.2012.08.008. PMID 22985997.
- ^ Hsueh YY, Chang YJ, Huang CW, Handayani F, Chiang YL, Fan SC, Ho CJ, Kuo YM, Yang SH, Chen YL, Lin SC, Huang CC, Wu CC (октябрь 2015 г.). «Синергия эндотелиальных и нервных клеток-предшественников из стволовых клеток, полученных из жировой ткани, для сохранения сосудисто-нервных структур при гипоксически-ишемическом повреждении головного мозга у крыс». Научные отчеты. 5: 14985. Bibcode:2015НатСР ... 514985H. Дои:10.1038 / srep14985. ЧВК 4597209. PMID 26447335.
- ^ Янив М. (сентябрь 2014 г.). «Ремоделирование хроматина: от транскрипции к раку». Генетика рака. 207 (9): 352–7. Дои:10.1016 / j.cancergen.2014.03.006. PMID 24825771.
- ^ Чжан Икс, Хе Н, Гу Д, Виклифф Дж, Салазар Дж, Болдог И., Се Дж (октябрь 2015 г.). «Генетические доказательства взаимодействий XPC-KRAS во время развития рака легких». Журнал генетики и геномики = И Чуань Сюэ Бао. 42 (10): 589–96. Дои:10.1016 / j.jgg.2015.09.006. ЧВК 4643398. PMID 26554912.
- ^ Дюбуа-Пот-Шнайдер Х, Фекир К., Кулуарн С., Глез Д., Анинат С., Ярнуен К., Ле Гевель Р., Кубо Т., Ишида С., Морель Ф, Корлу А. (декабрь 2014 г.). «Воспалительные цитокины способствуют ретродифференцировке опухолевых гепатоцитоподобных клеток в клетки-предшественники». Гепатология. 60 (6): 2077–90. Дои:10.1002 / hep.27353. PMID 25098666.
- ^ Финкин С., Юань Д., Стейн И., Танигучи К., Вебер А., Унгер К. и др. (Декабрь 2015 г.). «Эктопические лимфоидные структуры функционируют как микроники для клеток-предшественников опухоли при гепатоцеллюлярной карциноме». Иммунология природы. 16 (12): 1235–44. Дои:10.1038 / ni.3290. ЧВК 4653079. PMID 26502405.
- ^ а б Vlahopoulos SA, Cen O, Hengen N, Agan J, Moschovi M, Critselis E, Adamaki M, Bacopoulou F, Copland JA, Boldogh I., Karin M, Chrousos GP (август 2015 г.). «Динамический аберрантный NF-κB стимулирует туморогенез: новая модель, охватывающая микросреду». Отзывы о цитокинах и факторах роста. 26 (4): 389–403. Дои:10.1016 / j.cytogfr.2015.06.001. ЧВК 4526340. PMID 26119834.
- ^ Гривенников С.И., Карин М. (февраль 2010). «Опасные связи: сотрудничество STAT3 и NF-kappaB и перекрестные помехи при раке». Отзывы о цитокинах и факторах роста. 21 (1): 11–9. Дои:10.1016 / j.cytogfr.2009.11.005. ЧВК 2834864. PMID 20018552.
- ^ Ригер С., Чжао Х., Мартин П., Абе К., Лиссе Т.С. (январь 2015 г.). «Роль рецепторов ядерных гормонов в заживлении кожных ран». Биохимия и функции клетки. 33 (1): 1–13. Дои:10.1002 / cbf.3086. ЧВК 4357276. PMID 25529612.
- ^ Лу Х, Ярбро РГ (февраль 2015 г.). «Отрицательная регуляция фосфорилирования RelA: новые игроки и их роль в развитии рака». Отзывы о цитокинах и факторах роста. 26 (1): 7–13. Дои:10.1016 / j.cytogfr.2014.09.003. PMID 25438737.
- ^ Сионов Р.В., Фридлендер З.Г., Гранот З. (декабрь 2015 г.). «Многогранная роль нейтрофилов в микросреде опухоли». Микроокружение рака. 8 (3): 125–58. Дои:10.1007 / s12307-014-0147-5. ЧВК 4714999. PMID 24895166.
- ^ Вентури, Себастьяно (2011). «Эволюционное значение йода». Современная химическая биология. 5 (3): 155–162. Дои:10.2174/187231311796765012. ISSN 1872-3136.
- ^ Вентури S (2014). «Йод, ПНЖК и йодолипиды в здоровье и болезнях: эволюционная перспектива». Эволюция человека. 29 (1–3): 185–205. ISSN 0393-9375.
- ^ Уолш С.Дж., Люер Калифорния, Бодин А.Б., Смит Калифорния, Кокс Х.Л., Нойес Д.Р., Маура Г. (декабрь 2006 г.). «Иммунные клетки эластожаберных ветвей как источник новых ингибиторов опухолевых клеток: значение для общественного здравоохранения». Интегративная и сравнительная биология. 46 (6): 1072–1081. Дои:10.1093 / icb / icl041. ЧВК 2664222. PMID 19343108.
- ^ Фогельштейн Б., Кинцлер К.В. (август 2004 г.). «Раковые гены и пути, которые они контролируют». Природа Медицина. 10 (8): 789–99. Дои:10,1038 / нм1087. PMID 15286780.
- ^ Бренд KA, Hermfisse U (апрель 1997 г.). «Аэробный гликолиз пролиферирующими клетками: стратегия защиты от активных форм кислорода». Журнал FASEB. 11 (5): 388–95. Дои:10.1096 / fasebj.11.5.9141507. PMID 9141507.
- ^ Bos JL (сентябрь 1989 г.). «Онкогены ras при раке человека: обзор». Исследования рака. 49 (17): 4682–9. PMID 2547513.
- ^ Чанг Э. Х., Фурт МЭ, Сколник Э. М., Лоуи Д. Р. (июнь 1982 г.). «Онкогенная трансформация клеток млекопитающих, индуцированная нормальным человеческим геном, гомологичным онкогену вируса саркомы мышей Харви». Природа. 297 (5866): 479–83. Bibcode:1982Натура 297..479С. Дои:10.1038 / 297479a0. PMID 6283358.
- ^ Влахопулос С.А., Логотети С., Микас Д., Гиарика А., Горгулис В., Зумпурлис В. (апрель 2008 г.). «Роль АТФ-2 в онкогенезе». BioEssays. 30 (4): 314–27. Дои:10.1002 / bies.20734. PMID 18348191.
- ^ Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova O, Hurley PJ, Bunz F, Hwang PM (июнь 2006 г.). «p53 регулирует митохондриальное дыхание». Наука. 312 (5780): 1650–3. Bibcode:2006Научный ... 312.1650M. Дои:10.1126 / science.1126863. PMID 16728594.
- ^ Knudson AG (апрель 1971 г.). «Мутация и рак: статистическое исследование ретинобластомы». Труды Национальной академии наук Соединенных Штатов Америки. 68 (4): 820–3. Bibcode:1971ПНАС ... 68..820К. Дои:10.1073 / pnas.68.4.820. ЧВК 389051. PMID 5279523.
- ^ Фодде Р., Смитс Р. (октябрь 2002 г.). «Биология рака. Вопрос дозировки». Наука. 298 (5594): 761–3. Дои:10.1126 / science.1077707. PMID 12399571.
- ^ Стивенс П.Дж., Гринман С.Д., Фу Б., Янг Ф., Бигнелл Г.Р., Муди Л.Дж. и др. (Январь 2011 г.). «Массивная геномная перестройка, произошедшая в результате одного катастрофического события во время развития рака». Ячейка. 144 (1): 27–40. Дои:10.1016 / j.cell.2010.11.055. ЧВК 3065307. PMID 21215367. Сложить резюме – Нью-Йорк Таймс (10 января 2011 г.).
- ^ Kuipers EJ. Обзорная статья: изучение связи между Helicobacter pylori и раком желудка. Пищевая фармакология и терапия. Дополнение от марта 1999 г., т. 13, стр. 3-11. 9п. DOI: 10.1046 / j.1365-2036.1999.00002.x. (открытый доступ).
- ^ Кустерс Дж. Г., ван Влит А. Х., Кейперс Э. Дж. (Июль 2006 г.). «Патогенез инфекции Helicobacter pylori». Clin. Microbiol. Rev. 19 (3): 449–90. Дои:10.1128 / CMR.00054-05. ЧВК 1539101. PMID 16847081.
- ^ Паркин Д.М. (июнь 2006 г.). «Глобальное бремя рака, связанного с инфекциями, для здоровья в 2002 году». Int. J. Рак. 118 (12): 3030–44. Дои:10.1002 / ijc.21731. PMID 16404738.
- ^ Вроблевски Л. Е., Пик Р. М., Уилсон К. Т. (октябрь 2010 г.). «Helicobacter pylori и рак желудка: факторы, влияющие на риск заболевания». Clin. Microbiol. Rev. 23 (4): 713–39. Дои:10.1128 / CMR.00011-10. ЧВК 2952980. PMID 20930071.
- ^ Ферлей Дж., Коломбет М., Сурджоматарам И., Мазерс С., Паркин Д.М., Пиньерос М., Знаор А., Брей Ф. (апрель 2019 г.). «Оценка глобальной заболеваемости и смертности от рака в 2018 году: источники и методы GLOBOCAN». Int. J. Рак. 144 (8): 1941–1953. Дои:10.1002 / ijc.31937. PMID 30350310.
- ^ Meurer LN, Bower DJ (апрель 2002 г.). «Управление инфекцией Helicobacter pylori». Am Fam Врач. 65 (7): 1327–36. PMID 11996414.
- ^ Прабху С. Р., Ранганатан С., Амарапуркар Д. Н. (ноябрь 1994 г.). «Helicobacter pylori в нормальной слизистой оболочке желудка». J Assoc Physitors Индия. 42 (11): 863–4. PMID 7868485.
- ^ Белый JR, Винтер JA, Робинсон K (2015). «Дифференциальный воспалительный ответ на инфекцию Helicobacter pylori: этиология и клинические исходы». J Inflamm Res. 8: 137–47. Дои:10.2147 / JIR.S64888. ЧВК 4540215. PMID 26316793.
- ^ Дэн Дж.Й., Лян Х. (апрель 2014 г.). «Клиническое значение метастазов в лимфатические узлы при раке желудка». Мир J. Гастроэнтерол. 20 (14): 3967–75. Дои:10.3748 / wjg.v20.i14.3967. ЧВК 3983452. PMID 24744586.
- ^ а б Валенсуэла Массачусетс, Каналес Дж., Корвалан А.Х., Quest AF (декабрь 2015 г.). «Воспаление, вызванное Helicobacter pylori, и эпигенетические изменения во время канцерогенеза желудка». Мир J. Гастроэнтерол. 21 (45): 12742–56. Дои:10.3748 / wjg.v21.i45.12742. ЧВК 4671030. PMID 26668499.
- ^ Раза Й, Хан А., Фаруки А., Мубарак М., Фасиста А., Ахтар С. С., Хан С., Кази Дж. И., Бернштейн С., Казми С.У. (2014). «Окислительное повреждение ДНК как потенциальный ранний биомаркер канцерогенеза, связанного с Helicobacter pylori». Патол. Онкол. Res. 20 (4): 839–46. Дои:10.1007 / s12253-014-9762-1. PMID 24664859.
- ^ Коппель М., Гарсия-Алькальде Ф., Гловински Ф., Шлаерманн П., Мейер Т.Ф. (июнь 2015 г.). «Инфекция Helicobacter pylori вызывает характерные паттерны повреждения ДНК в клетках человека». Сотовый представитель. 11 (11): 1703–13. Дои:10.1016 / j.celrep.2015.05.030. PMID 26074077.
- ^ а б Мухаммад Дж. С., Эладль М. А., Ходер Г. (февраль 2019 г.). "Helicobacter pylori-индуцированное метилирование ДНК как эпигенетический модулятор рака желудка: недавние результаты и будущее направление". Патогены. 8 (1). Дои:10.3390 / pathogens8010023. ЧВК 6471032. PMID 30781778.
- ^ а б Ното Дж. М., Пик Р. М. (2011). «Роль микроРНК в патогенезе Helicobacter pylori и канцерогенезе желудка». Микробиол фронтальной клеточной инфекции. 1: 21. Дои:10.3389 / fcimb.2011.00021. ЧВК 3417373. PMID 22919587.
- ^ Сантос JC, Рибейро ML (август 2015 г.). «Эпигенетическая регуляция механизма репарации ДНК при канцерогенезе желудка, вызванном Helicobacter pylori». Мир J. Гастроэнтерол. 21 (30): 9021–37. Дои:10.3748 / wjg.v21.i30.9021. ЧВК 4533035. PMID 26290630.
- ^ а б Раза Й, Ахмед А., Хан А., Чишти А.А., Ахтер С.С., Мубарак М., Бернштейн С., Зайтлин Б., Казми С.У. (февраль 2020 г.). «Helicobacter pylori серьезно снижает экспрессию белков репарации ДНК PMS2 и ERCC1 при гастрите и раке желудка». Ремонт ДНК (Amst.). 89: 102836. Дои:10.1016 / j.dnarep.2020.102836. PMID 32143126.
- ^ Доре МП, Пес GM, Бассотти Дж., Усай-Сатта П. (2016). «Диспепсия: когда и как тестировать на инфекцию Helicobacter pylori». Гастроэнтерол Рес Прак. 2016: 8463614. Дои:10.1155/2016/8463614. ЧВК 4864555. PMID 27239194.
- ^ Каррильо-Инфанте С., Аббадесса Г., Багелла Л., Джордано А. (июнь 2007 г.). «Вирусные инфекции как причина рака (обзор)». Международный журнал онкологии. 30 (6): 1521–8. Дои:10.3892 / ijo.30.6.1521. PMID 17487374.
- ^ Сафдар А (2011). Лечение инфекций у онкологических больных. Springer. п. 478. ISBN 978-1-60761-643-6.
- ^ Самарас В., Рафаилидис П.И., Мурцуку Е.Г., Пеппас Г., Фалагас М.Э. (июнь 2010 г.). «Хронические бактериальные и паразитарные инфекции и рак: обзор». Журнал инфекций в развивающихся странах. 4 (5): 267–81. Дои:10.3855 / jidc.819. PMID 20539059.
- ^ Даниэль Ф.И., Керубини К., Юргель Л.С., де Фигейредо М.А., Салум Ф.Г. (февраль 2011 г.). «Роль эпигенетической репрессии транскрипции и ДНК-метилтрансфераз при раке». Рак. 117 (4): 677–87. Дои:10.1002 / cncr.25482. PMID 20945317. Обзор.
- ^ Канвал Р., Гупта С. (апрель 2012 г.). «Эпигенетические модификации рака». Клиническая генетика. 81 (4): 303–11. Дои:10.1111 / j.1399-0004.2011.01809.x. ЧВК 3590802. PMID 22082348.
- ^ Паттани К.М., Судри Э., Глейзер К.А., Охс М.Ф., Ван Х., Шуссель Дж., Сан В., Хеннесси П., Майдларц В., Лойо М., Демокан С., Смит И.М., Калифано Дж. А. (2012). Тао Q (ред.). «MAGEB2 активируется деметилированием промотора в плоскоклеточной карциноме головы и шеи». PLOS One. 7 (9): e45534. Bibcode:2012PLoSO ... 745534P. Дои:10.1371 / journal.pone.0045534. ЧВК 3454438. PMID 23029077.
- ^ Сампатх Д., Лю С., Васан К., Сульда М., Пудувалли В.К., Виерда В.Г., Китинг М.Дж. (февраль 2012 г.). «Гистоновые деацетилазы опосредуют подавление miR-15a, miR-16 и miR-29b при хроническом лимфоцитарном лейкозе». Кровь. 119 (5): 1162–72. Дои:10.1182 / кровь-2011-05-351510. ЧВК 3277352. PMID 22096249.
- ^ Хитчлер MJ, Оберли LW, Доманн FE (декабрь 2008 г.). «Эпигенетическое подавление SOD2 модификациями гистонов в клетках рака груди человека». Свободная радикальная биология и медицина. 45 (11): 1573–80. Дои:10.1016 / j.freeradbiomed.2008.09.005. ЧВК 2633123. PMID 18845242.
- ^ Baldassarre G, Battista S, Belletti B, Thakur S, Pentimalli F, Trapasso F, Fedele M, Pierantoni G, Croce CM, Fusco A (апрель 2003 г.). «Отрицательная регуляция экспрессии гена BRCA1 белками HMGA1 объясняет снижение уровней белка BRCA1 при спорадической карциноме молочной железы». Молекулярная и клеточная биология. 23 (7): 2225–38. Дои:10.1128 / MCB.23.7.2225-2238.2003. ЧВК 150734. PMID 12640109.
- ^ Далерба П., Чо Р. В., Кларк М. Ф. (2007). «Раковые стволовые клетки: модели и концепции». Ежегодный обзор медицины. 58: 267–84. Дои:10.1146 / annurev.med.58.062105.204854. PMID 17002552.
- ^ Чжан В., Хэнкс А.Н., Баучер К., Флорелл С.Р., Аллен С.М., Александр А., Браш Д.Е., Гроссман Д. (январь 2005 г.). «Апоптоз, вызванный УФ-В, стимулирует клональную экспансию во время развития опухоли кожи». Канцерогенез. 26 (1): 249–57. Дои:10.1093 / carcin / bgh300. ЧВК 2292404. PMID 15498793.
дальнейшее чтение
- Токар Э.Дж., Бенбрахим-Таллаа Л., Ваалкес МП (2011). "Chepter 14. Ионы металлов в развитии рака человека". В Астрид Сигель, Гельмут Сигель и Роланд К. О. Сигель (ред.). Ионы металлов в токсикологии: эффекты, взаимодействия, взаимозависимости. Ионы металлов в науках о жизни. 8. Издательство РСК. С. 375–401. Дои:10.1039/9781849732116-00375. ISBN 978-1-84973-091-4.
- Диксон К., Копрас Э. (декабрь 2004 г.). «Генетические изменения и репарация ДНК в канцерогенезе человека». Семинары по биологии рака. 14 (6): 441–8. Дои:10.1016 / j.semcancer.2004.06.007. PMID 15489137.
- Кляйнсмит LJ (2006). Принципы биологии рака. Сан-Франциско: Пирсон Бенджамин Каммингс. ISBN 978-0-8053-4003-7.
- Сарасин А. (ноябрь 2003 г.). «Обзор механизмов мутагенеза и канцерогенеза». Мутационные исследования. 544 (2–3): 99–106. Дои:10.1016 / j.mrrev.2003.06.024. PMID 14644312.
- Шоттенфельд Д., Бибе-Диммер Дж. Л. (2005). «Достижения в эпидемиологии рака: понимание причинно-следственных механизмов и данные для реализации вмешательств». Ежегодный обзор общественного здравоохранения. 26: 37–60. Дои:10.1146 / annurev.publhealth.26.021304.144402. PMID 15760280.
- Таннок I, Хилл Р., Бристоу Р., Харрингтон Л. (2005). Основная наука онкология (4-е изд.). Нью-Йорк: Макгроу-Хилл. ISBN 978-0-07-138774-3.
- Вича М.С., Лю С., Донту Г. (февраль 2006 г.). «Раковые стволовые клетки: старая идея - смена парадигмы». Исследования рака. 66 (4): 1883–90, обсуждение 1895–6. Дои:10.1158 / 0008-5472.CAN-05-3153. PMID 16488983.