Болезнь Альцгеймера с ранним началом - Википедия - Early-onset Alzheimers disease
| Болезнь Альцгеймера с ранним началом | |
|---|---|
| Специальность | Неврология |
Болезнь Альцгеймера с ранним началом, также называемый болезнь Альцгеймера с ранним началом, болезнь Альцгеймера с более ранним началом [1] или же раннее начало AD, является Болезнь Альцгеймера диагностирован в возрасте до 65 лет. Это необычная форма болезни Альцгеймера, на которую приходится всего 5–10% всех случаев болезни Альцгеймера. Около 60% имеют положительный семейный анамнез болезни Альцгеймера, и 13% из них наследуются по аутосомно-доминантному типу. Однако большинство случаев болезни Альцгеймера с ранним началом имеют те же черты, что и форма с «поздним началом», и не вызваны генетическими мутациями. Мало что известно о том, как это начинается.
Несемейная ранняя AD может развиться у людей в возрасте от 30 до 40 лет, но это происходит крайне редко.[2] Большинство людей с ранним началом болезни Альцгеймера находятся в возрасте от 50 до 60 лет.
История болезни Альцгеймера
Симптомы болезни как отчетливые нозологический сущность была впервые идентифицирована Эмиль Крепелин, а характерная невропатология была впервые обнаружена Алоис Альцгеймер в 1906 году. В этом смысле болезнь была открыта совместно Крепелином и Альцгеймером, которые работали в лаборатории Крепелина. Из-за огромной важности, которую Крепелин придавал обнаружению невропатологической основы психических расстройств, Крепелин принял решение, что болезнь будет носить имя Альцгеймера.[3]
Семейная болезнь Альцгеймера
Семейная болезнь Альцгеймера (FAD) или семейная болезнь Альцгеймера с ранним началом (EOFAD) - это необычная форма болезни Альцгеймера, которая обычно возникает в более раннем возрасте, определяется как до 65 лет (обычно в возрасте от 30 до 60 лет) и передается по наследству в аутосомно-доминантный мода, определяемая генетикой и другими характеристиками, такими как возраст начала. Семейная AD требует, чтобы у пациента был хотя бы один родственник первой степени с историей EOAD. FAD обычно подразумевает поражение нескольких человек в одном или нескольких поколениях.[4] Несемейные случаи БА называются «спорадическими», когда генетические факторы риска незначительны или неясны.[нужна цитата ]
В то время как семейная AD с ранним началом, по оценкам, составляет только 1% от общего числа случаев болезни Альцгеймера,[2] он представил полезную модель для изучения различных аспектов расстройства. В настоящее время мутации генов с ранним началом семейной болезни Альцгеймера определяют подавляющее большинство терапевтических открытий и разработок, основанных на моделях на животных, для лечения AD.[нужна цитата ]
Клинические признаки
Болезнь Альцгеймера (БА) - наиболее частая причина слабоумие и обычно происходит в старость. Это всегда приводит к летальному исходу, как правило, в течение 10 лет после появления первых признаков. Ранние признаки БА включают необычную потерю памяти, особенно при запоминании недавних событий и имен людей и вещей, логопеническая первично-прогрессирующая афазия. По мере прогрессирования болезни у пациента появляются более серьезные проблемы, он становится подверженным перепадам настроения и не может выполнять сложные действия, например, управлять автомобилем. К другим частым проявлениям относятся спутанность сознания, плохое суждение, нарушение речи, возбуждение, ломка, галлюцинации, судороги, симптомы болезни Паркинсона, повышенный мышечный тонус, миоклонус, недержание мочи и мутизм.[4] На последних стадиях они забывают, как делать простые вещи, например, расчесывать волосы, и им требуется постоянный уход.
Гистологически, семейная БА практически неотличима от других форм заболевания. Депозиты амилоид можно увидеть в разделах мозг ткань. Этот амилоидный белок образует бляшки и нейрофибриллярные сплетения что прогрессирует через мозг. Очень редко налет может быть уникальным или нехарактерным для БА; это может произойти, когда в одном из генов происходит мутация, которая создает функциональный, но уродливый белок вместо неэффективных генных продуктов, которые обычно возникают в результате мутаций.[нужна цитата ]
Нейробиология, лежащая в основе этого заболевания, только недавно начала изучаться. Исследователи работают над картированием путей воспаления, связанных с развитием, прогрессированием и дегенеративными свойствами AD. Основные молекулы, участвующие в этих путях, включают глиальные клетки (в частности, астроциты и микроглию), бета-амилоид и провоспалительные соединения. Поскольку нейроны повреждаются и умирают по всему мозгу, связи между сетями нейронов могут нарушаться, и многие области мозга сокращаются. На заключительных стадиях болезни Альцгеймера этот процесс, называемый атрофией мозга, становится широко распространенным, вызывая значительную потерю объема мозга. Эта потеря объема мозга влияет на способность жить и нормально функционировать и в конечном итоге приводит к летальному исходу.[5]
Бета-амилоид - это небольшой фрагмент более крупного белка, называемого белком-предшественником амилоида (APP). Как только APP активируется, он разрезается на более мелкие части других белков. Один из фрагментов, образующихся в процессе разрезания, - это β-амилоид. β-амилоид «более липкий», чем любой другой фрагмент, полученный из разрезанного APP, поэтому он запускает процесс накопления в мозге, который происходит из-за различных генетических и биохимических аномалий. В конце концов, фрагменты образуют олигомеры, затем фибриллы, бета-листы и, наконец, бляшки. Присутствие β-амилоидных бляшек в головном мозге заставляет организм рекрутировать и активировать микроглиальные клетки и астроциты.[нужна цитата ]
Генетика

Семейная болезнь Альцгеймера вызывается мутацией в одном из как минимум трех генов, которые кодируют пресенилин 1, пресенилин 2, и белок-предшественник амилоида (ПРИЛОЖЕНИЕ).[6][7][8] Другие генные мутации находятся в стадии изучения.
PSEN1 - Пресенилин 1
Ген пресенилина 1 (PSEN1 расположен на хромосоме 14) был идентифицирован Шеррингтоном (1995)[9] и были идентифицированы множественные мутации. Мутации в этом гене с уверенностью вызывают семейную болезнь Альцгеймера 3-го типа и обычно в возрасте до 50 лет. На этот тип приходится 30-70% EOFAD.[4] Этот белок был идентифицирован как часть ферментативного комплекса, который отщепляет бета-амилоидный пептид от АРР (см. Ниже).
Ген содержит 14 экзоны, а кодирующая часть оценивается в 60 кб, как сообщает Рогаев (1997).[10] и Дель-Фаверо (1999).[11] Белок, кодируемый геном (PS1), представляет собой интегральный мембранный белок. Как заявил Икеучи (2002)[12] он расщепляет белок Notch1, как полагает Коидзуми (2001)[13] играть роль в сомитогенезе у эмбриона. Он также действует на белок-предшественник амилоида, что дает его вероятную роль в патогенезе FAD. Гомологи PS1 были обнаружены у растений, беспозвоночных и других позвоночных.
Некоторые из мутаций в гене, из которых известно более 90, включают: His163Arg, Ala246Glu, Leu286Val и Cys410Tyr. Большая часть отображения завершена пенетрантность, но распространенной мутацией является Glu318Gly, и это предрасполагает людей к семейной БА, согласно исследованию Taddei (2002)[14] обнаруживая частоту 8,7% у пациентов с семейной АД.
PSEN2 - Пресенилин 2
Ген пресенилина 2 (PSEN2 ) очень похож по структуре и функциям на PSEN1. Он расположен на хромосоме 1 (1q31-q42), и мутации в этом гене вызывают FAD 4 типа. На этот тип приходится менее 5% всех случаев EOFAD.[4] Ген был идентифицирован Рудольфом Танци и Джерри Шелленбергом в 1995 году.[15] Последующее исследование Ковача (1996)[16] показали, что белки PS1 и PS2 экспрессируются в одинаковых количествах и в одинаковых органеллы как друг друга, в млекопитающее нейронный клетки. Леви-Лахад (1996)[17] определил, что PSEN2 содержит 12 экзонов, 10 из которых кодируют экзоны, и что первичный транскрипт кодирует 448-аминокислотный полипептид с 67% гомологией PS1. Этот белок был идентифицирован как часть ферментативного комплекса, который отщепляет бета-амилоидный пептид от АРР (см. Ниже).
Мутации изучены не так много, как PSEN1, но были идентифицированы различные аллельные варианты. К ним относятся Asn141Ile, который был впервые идентифицирован Рудольфом Танци и Джерри Шелленбергом в семьях поволжских немцев с семейной болезнью Альцгеймера (Levy-Lahad et al. Nature, 1995). Одно из этих исследований Ночлина (1998) обнаружило тяжелый амилоид ангиопатия у пострадавших в семье. Этот фенотип можно объяснить исследованием Томиты (1997).[18] предполагая, что мутация Asn141Ile изменяет метаболизм белка-предшественника амилоида (АРР), вызывая повышенную скорость отложения белка в бляшках.
Другими аллельными вариантами являются Met239Val, который был идентифицирован в итальянской родословной Рогаевым (1995).[19] которые также на раннем этапе предположили, что этот ген может быть похож на PSEN1, и мутацию Asp439Ala в экзоне 12 гена, которую предположил Lleo (2001)[20] для изменения эндопротеолитической обработки PS2.
APP - белок-предшественник бета (A4) амилоида
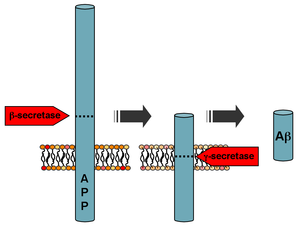
Мутации в белок-предшественник амилоида бета A4 (APP) расположенная на длинном плече хромосомы 21 (21q21.3), вызывает семейную болезнь Альцгеймера.[8]
[21] На этот вид приходится не более 10-15% EOFAD.[4]
Два разных ПРИЛОЖЕНИЕ выявленные и охарактеризованные мутации Шведский мутация[22] и Арктический мутация.[23] Функциональный анализ этих мутаций значительно расширил понимание патогенеза заболевания. В то время как Шведский Мутация, расположенная в сайте расщепления β-секретазой, приводит к общему более высокому производству пептидов Aβ за счет увеличения β-секреторного расщепления,[24] то Арктический Мутация приводит к изменению конформации пептида Aβ и повышенному образованию токсичных протофибрилл Aβ.[25]
Патофизиология
После расщепления β-секретаза, АРР расщепляется мембранно-связанным белковым комплексом, называемым γ-секретазой, с образованием Aβ.[26] Пресенилины 1 и 2 являются ферментативными центрами этого комплекса вместе с никастрином, Aph1 и PEN-2. Расщепление АРР альфа-секретазой, которое препятствует продукции Aβ, является наиболее частым событием процессинга АРР. В гене APP обнаружена 21 аллельная мутация. Они гарантируют раннее начало семейной болезни Альцгеймера, и все они происходят в области гена APP, кодирующего домен Aβ.
Генетическое тестирование
Генетическое тестирование доступно для лиц с симптомами и бессимптомных родственников.[7] Среди семей с EOFAD 40–80% будут иметь обнаруживаемую мутацию в гене APP, PSEN1 или PSEN2. Следовательно, в некоторых семьях с EOFAD не будет выявленной мутации при текущем тестировании.
Влияние болезни Альцгеймера с ранним началом
Нетипичное время протекания жизни для раннего начала болезни Альцгеймера означает, что она оказывает заметное влияние на переживания. Например, болезнь может иметь разрушительные последствия для карьеры, опекунов и членов семей пациентов.[27][28]
Те, кто работают, теряют способность выполнять свою работу компетентно и вынуждены досрочный выход на пенсию. Когда это можно предсказать, сотрудники должны обсудить свое будущее со своими работодателями и потерю навыков, с которыми они ожидают столкнуться.[29] Те, кто вынужден выходить на пенсию досрочно, могут не иметь доступа к полному спектру льгот, доступных тем, кто выходит на пенсию в минимальном возрасте, установленном государством.[29] В некоторых случаях ошибка может иметь разрушительные последствия для большого числа людей, и сообщалось о случаях, когда человек с ранним началом болезни Альцгеймера, не подозревающий о своем состоянии, вызвал страдания.[30][мертвая ссылка ]
Молодые люди, страдающие болезнью Альцгеймера, также могут потерять способность заботиться о своих потребностях, например о управлении деньгами.[31]
Однако также было подчеркнуто, что концептуальные представления о болезни Альцгеймера и старении должны сопротивляться представлению о существовании двух различных состояний.[32] Бинарная модель, которая сосредоточена, в частности, на потребностях молодых людей, может привести к тому, что проблемы, с которыми сталкиваются пожилые люди, будут недооценены.[33]
Смотрите также
- Еще Алиса (Роман) и фильм Еще Алиса, у главного героя которого есть EOAD
- Незабываемый дух, документальный фильм о прощальном турне музыканта Джон Манн и его группа Дух Запада после его диагноза с ранним началом болезни Альцгеймера
- Танматра (фильм), отмеченный наградами индийский фильм, в котором подробно рассказывается о влиянии болезни Альцгеймера на отца и его отношениях с сыном.
Рекомендации
- ^ «Болезнь Альцгеймера в более раннем возрасте / с ранним началом». Ассоциация Альцгеймера. Получено 9 июля 2020.
- ^ а б Харви Р.Дж., Скелтон-Робинсон М., Россор М.Н. (сентябрь 2003 г.). «Распространенность и причины деменции у людей в возрасте до 65 лет». Журнал неврологии, нейрохирургии и психиатрии. 74 (9): 1206–9. Дои:10.1136 / jnnp.74.9.1206. ЧВК 1738690. PMID 12933919.
- ^ Вебер М.М. (1997). «Алоис Альцгеймер, соратник Эмиля Крепелина». Журнал психиатрических исследований. 31 (6): 635–43. Дои:10.1016 / S0022-3956 (97) 00035-6. PMID 9447568.
- ^ а б c d е Bird, Thomas D. (1993), Adam, Margaret P .; Ardinger, Holly H .; Пагон, Роберта А .; Уоллес, Стефани Э. (ред.), «Семейная болезнь Альцгеймера с ранним началом - ГЛАВА В АРХИВЕ, ТОЛЬКО ДЛЯ ИСТОРИЧЕСКОЙ СПРАВКИ», GeneReviews®, Вашингтонский университет, Сиэтл, PMID 20301414, получено 2020-05-07
- ^ «Что происходит с мозгом при болезни Альцгеймера?». Национальный институт старения. Получено 2020-05-07.
- ^ Бертрам Л., Танзи Р. Э. (октябрь 2008 г.). «Тридцать лет генетики болезни Альцгеймера: последствия систематических метаанализов». Обзоры природы. Неврология. 9 (10): 768–78. Дои:10.1038 / номер 2494. PMID 18802446. S2CID 5946769.
- ^ а б Уильямсон Дж, Голдман Дж, Мардер К.С. (март 2009 г.). «Генетические аспекты болезни Альцгеймера». Невролог. 15 (2): 80–6. Дои:10.1097 / NRL.0b013e318187e76b. ЧВК 3052768. PMID 19276785.
- ^ а б Эртекин-Танер Н (август 2007 г.). «Генетика болезни Альцгеймера: столетний обзор». Неврологические клиники. 25 (3): 611–67, т. Дои:10.1016 / j.ncl.2007.03.009. ЧВК 2735049. PMID 17659183.
- ^ Шеррингтон Р., Рогаев Е.И., Лян Ю., Рогаева Е.А., Левеск Дж., Икеда М. и др. (Июнь 1995 г.). «Клонирование гена, несущего миссенс-мутации, при семейной болезни Альцгеймера с ранним началом». Природа. 375 (6534): 754–60. Bibcode:1995Натура.375..754S. Дои:10.1038 / 375754a0. PMID 7596406. S2CID 4308372.
- ^ Рогаев Е.И., Шеррингтон Р., Ву К., Левеск Дж., Лян Ю., Рогаева Е.А. и др. (Март 1997 г.). «Анализ 5'-последовательности, геномной структуры и альтернативного сплайсинга гена пресенилина-1 (PSEN1), связанного с ранним началом болезни Альцгеймера». Геномика. 40 (3): 415–24. Дои:10.1006 / geno.1996.4523. PMID 9073509.
- ^ Дель-Фаверо Дж., Гуссенс Д., Ван ден Босше Д., Ван Брокховен С. (март 1999 г.). «Фрагментация YAC с повторяющимися и однокопийными последовательностями: подробное физическое картирование гена пресенилина 1 на хромосоме 14». Ген. 229 (1–2): 193–201. Дои:10.1016 / S0378-1119 (99) 00023-2. PMID 10095119.
- ^ Икеучи Т., Сисодиа СС (2002). «Бесклеточная генерация внутриклеточного домена notch1 (NICD) и APP-CTfgamma: доказательства различных внутримембранных активностей« гамма-секретазы »». Нейромолекулярная медицина. 1 (1): 43–54. Дои:10.1385 / НММ: 1: 1: 43. PMID 12025815. S2CID 21552663.
- ^ Коидзуми К., Накадзима М., Юаса С., Сага Ю., Сакаи Т., Курияма Т. и др. (Апрель 2001 г.). «Роль пресенилина 1 при сегментации сомитов». Разработка. 128 (8): 1391–402. PMID 11262239.
- ^ Таддей К., Фишер С., Лоус С.М., Мартинс Г., Патон А., Кларнетт Р.М. и др. (2002). «Связь между мутацией пресенилина-1 Glu318Gly и семейной болезнью Альцгеймера у населения Австралии». Молекулярная психиатрия. 7 (7): 776–81. Дои:10.1038 / sj.mp.4001072. PMID 12192622.
- ^ Леви-Лахад Э., Васко В., Пуркадж П., Романо Д.М., Осима Дж., Петтингелл У.Х. и др. (Август 1995 г.). «Ген-кандидат в семейный локус болезни Альцгеймера на хромосоме 1». Наука. 269 (5226): 973–7. Bibcode:1995Научный ... 269..973L. Дои:10.1126 / science.7638622. PMID 7638622.
- ^ Ковач Д.М., Фосетт Х.Дж., Пейдж К.Дж., Ким Т.В., Мойр Р.Д., Мерриам Д.Э. и др. (Февраль 1996 г.). «Связанные с болезнью Альцгеймера пресенилины 1 и 2: экспрессия нейронов в головном мозге и локализация на внутриклеточных мембранах в клетках млекопитающих». Природа Медицина. 2 (2): 224–9. Дои:10,1038 / нм0296-224. PMID 8574969. S2CID 25596140.
- ^ Леви-Лахад Э, Пооркадж П., Ван К., Фу Й.Х., Осима Дж., Маллиган Дж., Шелленберг Г.Д. (июнь 1996 г.). «Геномная структура и экспрессия STM2, хромосомы 1 семейного гена болезни Альцгеймера». Геномика. 34 (2): 198–204. Дои:10.1006 / geno.1996.0266. PMID 8661049.
- ^ Томита Т., Маруяма К., Сайдо Т.С., Куме Х., Шинозаки К., Токухиро С. и др. (Март 1997 г.). «Мутация пресенилина 2 (N141I), связанная с семейной болезнью Альцгеймера (семьи поволжских немцев), увеличивает секрецию бета-амилоидного белка, оканчивающегося на 42-м (или 43-м) остатке». Труды Национальной академии наук Соединенных Штатов Америки. 94 (5): 2025–30. Bibcode:1997PNAS ... 94.2025T. Дои:10.1073 / пнас.94.5.2025. JSTOR 41579. ЧВК 20036. PMID 9050898.
- ^ Рогаев Е.И., Шеррингтон Р., Рогаева Е.А., Левеск Дж., Икеда М., Лян Ю. и др. (Август 1995 г.). «Семейная болезнь Альцгеймера в родственниках с миссенс-мутациями в гене на хромосоме 1, связанном с геном болезни Альцгеймера 3 типа». Природа. 376 (6543): 775–8. Bibcode:1995Натура 376..775р. Дои:10.1038 / 376775a0. PMID 7651536. S2CID 4259326.
- ^ Ллео А., Блеса Р., Жендре Дж., Кастельви М., Пастор П., Керальт Р., Олива Р. (ноябрь 2001 г.). «Новая мутация гена пресенилина 2 (D439A) у пациента с ранним началом болезни Альцгеймера». Неврология. 57 (10): 1926–8. Дои:10.1212 / WNL.57.10.1926. PMID 11723295.
- ^ Маленка Э.Дж., Нестлер С.Е., Хайман Р.К. (2009). Молекулярная нейрофармакология: основа клинической неврологии (2-е изд.). Нью-Йорк: McGraw-Hill Medical. ISBN 9780071481274.[страница нужна ]
- ^ Муллан М., Кроуфорд Ф., Аксельман К., Хоулден Х., Лилиус Л., Винблад Б., Ланнфельт Л. (август 1992 г.). «Патогенная мутация вероятной болезни Альцгеймера в гене APP на N-конце бета-амилоида». Природа Генетика. 1 (5): 345–7. Дои:10.1038 / ng0892-345. PMID 1302033. S2CID 20046036.
- ^ Nilsberth C, Westlind-Danielsson A, Eckman CB, Condron MM, Axelman K, Forsell C и др. (Сентябрь 2001 г.). «Арктическая мутация APP (E693G) вызывает болезнь Альцгеймера за счет усиленного образования протофибрилл Abeta» (PDF). Природа Неврология. 4 (9): 887–93. Дои:10.1038 / nn0901-887. PMID 11528419.
- ^ Джонстон Дж. А., Кауберн Р. Ф., Норгрен С., Вихагер Б., Венизелос Н., Винблад Б. и др. (Ноябрь 1994 г.). «Повышенное высвобождение бета-амилоида и уровни белка-предшественника амилоида (APP) в клеточных линиях фибробластов от членов семьи с мутацией шведской болезни Альцгеймера APP670 / 671». Письма FEBS. 354 (3): 274–8. Дои:10.1016/0014-5793(94)01137-0. PMID 7957938.
- ^ Johansson AS, Berglind-Dehlin F, Karlsson G, Edwards K, Gellerfors P, Lannfelt L (июнь 2006 г.). «Физиохимическая характеристика пептидов, связанных с болезнью Альцгеймера, A beta 1-42Arctic и A beta 1-42wt». Журнал FEBS. 273 (12): 2618–30. Дои:10.1111 / j.1742-4658.2006.05263.x. PMID 16817891.
- ^ Чоу В.В., Маттсон М.П., Вонг ПК, Гляйхманн М. (март 2010 г.). «Обзор ферментов и продуктов для обработки APP». Нейромолекулярная медицина. 12 (1): 1–12. Дои:10.1007 / s12017-009-8104-z. ЧВК 2889200. PMID 20232515.
- ^ Сотрудники клиники Мэйо, Раннее начало болезни Альцгеймера: симптомы начинаются до 65 лет., Клиника Майо
- ^ Мэри Брофи Маркус, Семья разделяет путь после раннего диагноза Альцгеймера, USA Today (2 сентября 2008 г.).
- ^ а б Жизнь с болезнью Альцгеймера с ранним началом В архиве 2007-10-19 на Wayback Machine, Система здравоохранения клиники Кливленда
- ^ Раннее начало болезни Альцгеймера на подъеме, CBS Новости (8 марта 2008 г.).
- ^ Кэтлин Факельманн, Кто думает о болезни Альцгеймера у такого молодого человека?, USA Today (11 июня 2007 г.).
- ^ Рахман, С. (2016). Молодое слабоумие: слишком далеко?, Общество деменции, 27 июля 2016 г.
- ^ Толхерст Э (2016). «Растущий интерес к деменции у молодых: восстановление баланса или усиление эйджизма?» (PDF). Международный журнал старения и дальнейшей жизни. 10 (2): 9–29. Дои:10.3384 / ijal.1652-8670.16302.
внешняя ссылка
| Классификация |
|---|
- Семейная болезнь Альцгеймера с ранним началом - Томас Д. Берд, доктор медицины GeneRevies (Национальные институты здравоохранения США.gov)