Кислоты и основания Льюиса - Lewis acids and bases


А Кислота Льюиса это химический вид, содержащий пустой орбитальный который способен принять электронная пара от Льюиса основание сформировать Льюис аддукт. А База Льюиса, то есть любой вид, у которого есть заполненная орбиталь, содержащая электронная пара который не участвует в связь но может образовать дательная облигация с кислотой Льюиса с образованием аддукта Льюиса. Например, NH3 это база Льюиса, потому что она может пожертвовать одинокая пара электронов. Триметилборана (Мне3B) является кислотой Льюиса, поскольку она способна принимать неподеленную пару. В аддукте Льюиса кислота и основание Льюиса имеют общую электронную пару, предоставленную основанием Льюиса, образуя дательную связь.[1] В контексте конкретной химическая реакция между NH3 и я3B, одинокая пара из NH3 образует дательную связь с пустой орбиталью Меня3B с образованием аддукта NH3• BMe3. Терминология относится к вкладам Гилберт Н. Льюис.[2]
Условия нуклеофил и электрофил более или менее взаимозаменяемы с основанием Льюиса и кислотой Льюиса соответственно. Однако эти термины, особенно их абстрактные формы существительных нуклеофильность и электрофильность, подчеркивают кинетический аспект реакционной способности, в то время как основность Льюиса и кислотность Льюиса подчеркивают термодинамический аспект образования аддукта Льюиса.[3]
Изображение аддуктов
Во многих случаях взаимодействие между основанием Льюиса и кислотой Льюиса в комплексе показано стрелкой, указывающей на то, что основание Льюиса отдает электроны в сторону кислоты Льюиса, используя обозначение дательная облигация -Например, Мне3B ← NH3. Некоторые источники указывают на основание Льюиса парой точек (явные передаваемые электроны), что позволяет согласованно представить переход от самого основания к комплексу с кислотой:
- Мне3B +: NH3 → Мне3B: NH3
Центральная точка также может использоваться для обозначения аддукта Льюиса, такого как Мне3B • NH3. Другой пример диэтилэфират трифторида бора, BF3• Et2О. (В несколько ином использовании центральная точка также используется для обозначения гидратная координация в различных кристаллах, как в MgSO4• 7H2O для гидратированного сульфат магния, независимо от того, образует ли вода дативную связь с металлом.)
Хотя были попытки использовать вычислительные и экспериментальные энергетические критерии, чтобы отличить дативную связь от недативной ковалентной связи,[4] по большей части, различие просто указывает на источник электронной пары, и однажды образованные дативные связи ведут себя так же, как и другие ковалентные связи, хотя обычно они имеют значительный полярный характер. Более того, в некоторых случаях (например, сульфоксиды и оксиды аминов как R2S → O и R3N → O), использование стрелки дательного падежа - просто удобство обозначений, позволяющее избежать формальных сборов. В целом, однако, донорно-акцепторная связь рассматривается просто как нечто среднее между идеализированными ковалентная связь и ионная связь.[5]
Примеры
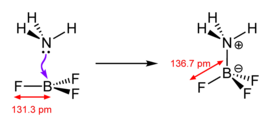
Обычно термин «кислота Льюиса» ограничивается тригонально плоский виды с пустой p-орбиталью, такие как BR3 где R может быть органическим заместителем или галогенидом.[нужна цитата ] Для целей обсуждения даже сложные соединения, такие как Et3Al2Cl3 и AlCl3 рассматриваются как тригональные планарные кислоты Льюиса. Ионы металлов, такие как Na+, Mg2+, а Ce3+, которые неизменно осложняются дополнительными лиганды, часто являются источниками координационно ненасыщенный производные, образующие Льюис аддукты после реакции с основанием Льюиса.[нужна цитата ] Другие реакции можно просто назвать реакциями, катализируемыми кислотой. Некоторые соединения, такие как H2O являются как кислотами Льюиса, так и основаниями Льюиса, потому что они могут либо принимать пару электронов, либо отдавать пару электронов, в зависимости от реакции.
Кислоты Льюиса разнообразны. Самыми простыми являются те, которые напрямую реагируют с базой Льюиса. Но чаще встречаются те, которые подвергаются реакции до образования аддукта.
- Примеры кислот Льюиса, основанные на общем определении акцептора электронной пары, включают:
- протон (H+) и кислотные соединения ионы ония, Такие как NH4+ и ЧАС3О+
- катионы переходных металлов с высокой степенью окисления, например, Fe3+;
- другие катионы металлов, такие как Ли+ и Mg2+, часто как их акво или эфирные комплексы,
- тригональные плоские виды, такие как BF3 и карбокатионы ЧАС3C+
- пентагалогениды фосфора, мышьяка и сурьмы
- электронная бедность π-системы, такие как Enones и тетрацианоэтилены.
Опять же, описание кислоты Льюиса часто используется нечетко. Например, в растворе голых протонов не существует.
Простые кислоты Льюиса
Некоторые из наиболее изученных примеров таких кислот Льюиса - это тригалогениды бора и органобораны, но другие соединения демонстрируют такое поведение:
- BF3 + F− → BF4−
В этом аддукте все четыре фторидных центра (точнее, лиганды ) эквивалентны.
- BF3 + OMe2 → BF3OMe2
Оба BF4− и BF3OMe2 являются аддуктами основания Льюиса трифторида бора.
Во многих случаях аддукты нарушают Правило октета, такой как трииодид анион:
- я2 + Я− → я3−
Вариабельность окраски растворов йода отражает переменную способность растворителя образовывать аддукты с кислотой Льюиса I.2.
В некоторых случаях кислота Льюиса способна связывать два основания Льюиса, известным примером является образование гексафторсиликат:
- SiF4 + 2 Ж− → SiF62−
Комплексные кислоты Льюиса
Большинство соединений, которые считаются кислотами Льюиса, требуют стадии активации перед образованием аддукта с основанием Льюиса. Хорошо известны тригалогениды алюминия, которые широко рассматриваются как кислоты Льюиса. Тригалогениды алюминия, в отличие от тригалогенидов бора, не существуют в форме AlX3, но как агрегаты и полимеры, которые должны разрушаться основанием Льюиса.[6] Более простой случай - образование аддуктов борана. Мономерная BH3 не существует в значительной степени, поэтому аддукты борана образуются в результате разложения диборана:
- B2ЧАС6 + 2 часа− → 2 ч.4−
В этом случае промежуточный B2ЧАС7− можно изолировать.
Многие комплексы металлов служат кислотами Льюиса, но обычно только после диссоциации более слабосвязанного основания Льюиса, часто воды.
- [Mg (H2O)6]2+ + 6 NH3 → [Mg (NH3)6]2+ + 6 часов2О
ЧАС+ как кислота Льюиса
В протон (ЧАС+) [7] является одной из самых сильных, но также и одной из самых сложных кислот Льюиса. Принято игнорировать тот факт, что протон сильно сольватирован (связан с растворителем). Имея в виду это упрощение, кислотно-основные реакции можно рассматривать как образование аддуктов:
- ЧАС+ + NH3 → NH4+
- ЧАС+ + ОН− → H2О
Применение кислот Льюиса
Типичный пример действия кислоты Льюиса находится в Алкилирование Фриделя – Крафтса реакция.[5] Ключевым шагом является принятие AlCl3 неподеленной пары хлорид-иона, образуя AlCl4− и создавая сильно кислый, то есть электрофильный, ион карбония.
- RCl + AlCl3 → R+ + AlCl4−
Базы Льюиса
Основание Льюиса - это атомарный или молекулярный вид, в котором самая высокая занятая молекулярная орбиталь (HOMO) сильно локализован. Типичные базы Льюиса - обычные амины такие как аммиак и алкил амины. Другие распространенные основания Льюиса включают пиридин и его производные. Некоторые из основных классов базисов Льюиса:
- амины формулы NH3−ИксрИкс где R = алкил или арил. К ним относятся пиридин и его производные.
- фосфины формулы PR3−ИксАИкс, где R = алкил, A = арил.
- соединения O, S, Se и Te в степени окисления -2, включая воду, эфиры, кетоны
Наиболее распространенными основаниями Льюиса являются анионы. Сила основности Льюиса коррелирует с pKа исходной кислоты: кислоты с высоким pKаДадим хорошие базы Льюиса. Как обычно, более слабая кислота имеет более сильный сопряженное основание.
- Примеры оснований Льюиса, основанных на общем определении донора электронной пары, включают:
Сила оснований Льюиса была оценена для различных кислот Льюиса, таких как I2, SbCl5, и BF3.[8]
| База Льюиса | Донор атом | Энтальпия комплексообразования (кДж / моль) |
|---|---|---|
| Et3N | N | 135 |
| хинуклидин | N | 150 |
| пиридин | N | 128 |
| Ацетонитрил | N | 60 |
| Et2О | О | 78.8 |
| THF | О | 90.4 |
| ацетон | О | 76.0 |
| EtOAc | О | 75.5 |
| DMA | О | 112 |
| ДМСО | О | 105 |
| Тетрагидротиофен | S | 51.6 |
| Триметилфосфин | п | 97.3 |
Приложения баз Льюиса
Почти все доноры электронных пар, которые образуют соединения путем связывания переходных элементов, можно рассматривать как совокупность оснований Льюиса - или лиганды. Таким образом, основное применение оснований Льюиса заключается в изменении активности и селективности металлических катализаторов. Таким образом, хиральные основания Льюиса дают хиральность на катализаторе, позволяющем асимметричный катализ, что полезно для производства фармацевтические препараты.
Многие основания Льюиса являются «мультидентатными», то есть они могут образовывать несколько связей с кислотой Льюиса. Эти мультидентатные основания Льюиса называются хелатирующие агенты.
Жесткая и мягкая классификация
Кислоты и основания Льюиса обычно классифицируются по их твердости или мягкости. В этом контексте жесткий означает малые и неполяризуемые, а мягкий означает более поляризуемые атомы большего размера.
- типичные твердые кислоты: H+, катионы щелочных / щелочноземельных металлов, бораны, Zn2+
- типичные мягкие кислоты: Ag+, Mo (0), Ni (0), Pt2+
- типичные твердые основания: аммиак и амины, вода, карбоксилаты, фторид и хлорид
- типичные мягкие основания: органофосфины, тиоэфиры, оксид углерода, йодид
Например, амин вытеснит фосфин из аддукта с кислотой BF3. Таким же образом можно было классифицировать базы. Например, основания, передающие неподеленную пару из атома кислорода, сложнее, чем основания, передающие через атом азота. Хотя классификация никогда не подвергалась количественной оценке, она оказалась очень полезной для прогнозирования силы образования аддукта, используя ключевые концепции, согласно которым взаимодействия твердой кислоты с твердым основанием и мягкой кислотой с мягким основанием сильнее, чем взаимодействия твердой кислоты с мягким основанием или мягкой кислотой с твердым. базовые взаимодействия. Более позднее исследование термодинамики взаимодействия показало, что жесткие и жесткие взаимодействия являются энтальпия предпочтительны, тогда как мягкие - мягкие энтропия одобренный.
Количественная оценка кислотности Льюиса
Для оценки и прогнозирования кислотности Льюиса было разработано множество методов. Многие из них основаны на спектроскопических сигнатурах, например сдвиги в 31Сигналы P ЯМР или ИК-диапазоны. Включены методы, разработанные Гутманном, Чайлдсом,[9] и Беккет.
В Модель ECW представляет собой количественную модель, которая описывает и предсказывает силу взаимодействия кислот Льюиса с основанием, -ΔH. Модель присвоила параметры E и C многим кислотам и основаниям Льюиса. Каждая кислота характеризуется буквой EА и CА. Каждой базе также соответствует свой EB и CB. Параметры E и C относятся, соответственно, к электростатическому и ковалентному вкладу в силу связей, которые будут образовывать кислота и основание. Уравнение
- −ΔH = EАEB + CАCB + W
Термин W представляет собой постоянный вклад энергии для кислотно-основной реакции, такой как расщепление димерной кислоты или основания. Уравнение предсказывает изменение силы кислот и оснований. Графическое представление уравнения показывает, что не существует единого порядка концентраций оснований Льюиса или кислот Льюиса.[10]
Не существует единого порядка концентраций оснований Льюиса или концентраций кислоты Льюиса.
Графики Крамера – Боппа показывают графически с использованием параметров E и C Модель ECW что не существует единого порядка концентраций оснований Льюиса (или кислотных сил).[11] Одно свойство или переменные масштабы ограничены небольшим диапазоном кислот или оснований.
История
Концепция возникла с Гилберт Н. Льюис кто учился химическая связь. В 1923 году Льюис написал Кислотное вещество - это вещество, которое может использовать неподеленную пару электронов из другой молекулы для завершения стабильной группы одного из своих собственных атомов.[2][12] В Кислотно-основная теория Бренстеда – Лоури вышла в том же году. Две теории различны, но дополняют друг друга. Основание Льюиса также является основанием Бренстеда-Лоури, но кислота Льюиса не обязательно должна быть кислотой Бренстеда-Лоури. Классификация на жесткие и мягкие кислоты и основания (Теория HSAB ) последовал в 1963 году. Сила кислотно-основных взаимодействий Льюиса, измеренная стандартом энтальпия образование аддукта можно предсказать с помощью двухпараметрического уравнения Драго – Вейланда.
Переформулировка теории Льюиса
Льюис предположил в 1916 г., что два атомы удерживаются вместе в химической связи, разделяя пару электронов.[13] Когда каждый атом вносил один электрон в связь, это называлось Ковалентная связь. Когда оба электрона исходят от одного из атомов, это называлось дательной ковалентной связью или координационная связь. Различие не очень четкое. Например, при образовании иона аммония из аммиака и водорода аммиак молекула отдает пару электронов протон;[7] идентичность электронов теряется в аммоний ион, который образуется. Тем не менее, Льюис предложил классифицировать донор электронной пары как основание, а акцептор электронной пары - как кислоту.
Более современное определение кислоты Льюиса - это атомарный или молекулярный вид с локализованной пустой атомный или же молекулярный орбиталь низкой энергии. Эта молекулярная орбиталь с наименьшей энергией (LUMO ) может вместить пару электронов.
Сравнение с теорией Бренстеда – Лоури.
База Льюиса часто является базой Бренстеда – Лоури, поскольку она может отдавать пару электронов H+;[7] протон является кислотой Льюиса, поскольку он может принимать пару электронов. Конъюгированное основание кислоты Бренстеда-Лоури также является основанием Льюиса, поскольку потеря H+ из кислоты уходит те электроны, которые использовались для связи A — H, как неподеленная пара на сопряженном основании. Однако базу Льюиса может быть очень сложно протонировать, но все еще реагирует с кислотой Льюиса. Например, монооксид углерода является очень слабым основанием Бренстеда – Лоури, но образует сильный аддукт с BF3.
В другом сравнении кислотности Льюиса и Бренстеда – Лоури, проведенного Брауном и Каннером,[14] 2,6-ди-т-бутилпиридин реагирует с образованием гидрохлоридной соли с HCl, но не реагирует с BF3. Этот пример демонстрирует, что стерические факторы, в дополнение к факторам электронной конфигурации, играют роль в определении силы взаимодействия между громоздкими ди-т-бутилпиридин и крошечный протон.
Смотрите также
- Кислота
- База (химия)
- Кислотно-основная реакция
- Кислотно-основная теория Бренстеда – Лоури
- Хиральная кислота Льюиса
- Разочарованная пара Льюиса
- Метод Гутмана – Беккета
- Модель ECW
Рекомендации
- ^ Золотая книга ИЮПАК - кислота Льюиса
- ^ а б Льюис, Гилберт Ньютон (1923). Валентность и структура атомов и молекул.. Нью-Йорк, Нью-Йорк, США: Компания по каталогу химических веществ. п. 142. С п. 142: «Мы склонны думать о веществах как обладающих кислотными или основными свойствами, не имея в виду конкретный растворитель. Мне кажется, что с полной общностью мы можем сказать, что основное вещество - это вещество, которое имеет неподеленную пару электронов, которая может быть использована для завершения стабильной группы другого атома, и это кислотное вещество - это вещество, которое может использовать неподеленную пару из другой молекулы в завершении стабильной группы одного из собственных атомов. Другими словами, основное вещество обеспечивает пару электронов для химической связи, кислотное вещество принимает такую пару ».
- ^ 1960-, Анслин, Эрик В. (2006). Современная физическая органическая химия. Догерти, Деннис А., 1952-. Саусалито, Калифорния: Университетская наука. ISBN 1891389319. OCLC 55600610.CS1 maint: числовые имена: список авторов (связь)
- ^ Лепетит, Кристина; Мараваль, Валери; Канак, Ив; Шовен, Реми (2016). «О природе дательной связи: координация с металлами и не только. Углеродный случай». Обзоры координационной химии. 308: 59–75. Дои:10.1016 / j.ccr.2015.07.018.
- ^ а б Марч, Дж. «Продвинутая органическая химия», 4-е изд. Дж. Вили и сыновья, 1992: Нью-Йорк. ISBN 0-471-60180-2.
- ^ Greenwood, N. N .; И Эрншоу, А. (1997). Химия элементов (2-е изд.), Оксфорд: Баттерворт-Хайнеманн. ISBN 0-7506-3365-4.
- ^ а б c Традиционно, но не точно, ЧАС+ ионы называются "протоны ". Видеть ИЮПАК, Сборник химической терминологии, 2-е изд. («Золотая книга») (1997). Исправленная онлайн-версия: (2006–) "гидрон ". Дои:10.1351 / goldbook.H02904
- ^ Кристиан Лоуренс и Жан-Франсуа Галь "Шкалы основности и сродства Льюиса: данные и измерения", Wiley, 2009. ISBN 978-0-470-74957-9.
- ^ Чайлдс, Р.Ф .; Mulholland, D.L; Никсон, А. (1982). «Аддукты кислоты Льюиса α, β-ненасыщенных карбонильных и нитрильных соединений. Исследование ядерного магнитного резонанса». Может. J. Chem. 60 (6): 801–808. Дои:10.1139 / v82-117.
- ^ Фогель Г. К.; Драго Р. С. (1996). «Модель ECW». Журнал химического образования. 73: 701–707. Bibcode:1996JChEd..73..701V. Дои:10.1021 / ed073p701.CS1 maint: использует параметр авторов (связь)
- ^ . Крамер Р. Э. и Бопп Т. Т. (1977) The Great E&C Plot. Графическое отображение энтальпий образования аддуктов для кислот и оснований Льюиса. Журнал химического образования 54 612–613.
- ^ Мисслер Л. М., Тар Д. А. (1991) стр. 166 - В таблице открытий датой публикации / выпуска теории Льюиса считается 1923 год.
- ^ Льюис, Гилберт Н. (апрель 1916 г.). «Атом и молекула». Журнал Американского химического общества. 38 (4): 762–785. Дои:10.1021 / ja02261a002.
- ^ Браун Х.С. и Каннер Б. "Получение и реакции 2,6-ди-т-бутилпиридин и родственные затрудненные основания. Случай стерического препятствия для протона ». J. Am. Chem. Soc. 88, 986 (1966).
дальнейшее чтение
- Дженсен, У.Б. (1980). Кислотно-основные концепции Льюиса: обзор. Нью-Йорк: Вили. ISBN 0-471-03902-0.
- Ямамото, Хисаши (1999). Реактивы кислоты Льюиса: практический подход. Нью-Йорк: Издательство Оксфордского университета. ISBN 0-19-850099-8.