Глутаматдегидрогеназа 1 - Glutamate dehydrogenase 1
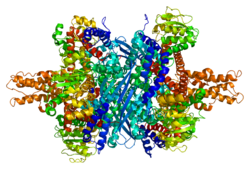





GLUD1 (глутаматдегидрогеназа 1) это митохондриальный матрица фермент, один из семьи глутаматдегидрогеназы которые вездесущи в жизнь, с ключевой ролью в азот и глутамат (Glu) метаболизм и энергия гомеостаз. Этот дегидрогеназа выражается на высоком уровне в печень, мозг, поджелудочная железа и почка, но не в мышца. В поджелудочной клетки, Предполагается, что GLUD1 участвует в инсулин секреция механизмы. В нервной ткани, где глутамат присутствует в концентрациях выше, чем в других тканях, GLUD1, по-видимому, действует как в синтез и катаболизм глутамата и, возможно, в аммиак детоксикация.
Структура
Ген
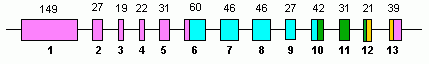
Человек GLUD1 содержит 13 экзоны и находится на 10-м хромосома.
Есть свидетельства того, что GLUD1 был перенесен в Х-хромосому, где он дал начало безинтронному GLUD2 через случайный мутации и естественный отбор. GLUD2 адаптировались к особым потребностям нервной системы, где это особенно выражено.[5]
Протеин

Каждый домен окрашен по-разному - Glu-BD, НАД (П) -БД, антенна, поворотная спираль. Аллостерические регуляторы показаны в виде сферических моделей. Эта конкретная структура GLUD1 представляет собой комбинацию двух рентгеновских структур - одна со связанным GTP (1HWZ ) и второй со связанным ADP (1NQT ). Хотя это и нереально, эта структура показывает относительное положение аллостерических эффекторов, когда они связаны с GLUD1. Также показаны NADPH и Glu.
GLUD1 - гексамер. В состав мономерного агрегата входят:
- N-концевой Glu-BD (связывающий домен), который состоит в основном из β-цепей.
- НАД-БД - может связывать либо НАД+ или НАДФ+.
- 48-остаточный антенно-подобный выступ, выходящий из верхней части каждого NAD-BD. Антенна состоит из восходящей спирали и нисходящей случайной нити катушки, которая содержит небольшую α-спираль к С-концевому концу нити.
NAD-BD находится на вершине Glu-BD. NAD-BD и Glu-BD образуют каталитическую щель. Во время связывания субстрата NAD-BD значительно перемещается. Это движение состоит из двух компонентов: они вращаются вдоль длинной оси спирали в задней части NAD-BD, называемой «поворотной спиралью», и вращаются вокруг антенны по часовой стрелке. Сравнение открытой и закрытой конформаций GLUD1 обнаруживает изменения в маленькой спирали нисходящей нити антенны, которая, кажется, отскакивает, когда открывается каталитическая щель.[6] Закрытие одной субъединицы связано с искажением небольшой спирали нисходящей нити, которая проталкивается в антенну соседней субъединицы. R496 расположен на этой маленькой спирали (см. Мутации).
Ядро гексамера представляет собой сложенный димер тримеров. Glu-BD мономеров в основном ответственны за наращивание ядра. Относительное положение мономеров таково, что вращение вокруг поворотной спирали в каждом мономере не ограничено. Антенны из трех субъединиц внутри тримеров обвиваются вокруг друг друга и претерпевают конформационные изменения, когда каталитическая щель открывается и закрывается. Антенна служит каналом связи между подразделениями во время отрицательной кооперативности и аллостерической регуляции.
Выравнивание GLUD1 из различных источников показывает, что антенна, вероятно, эволюционировала у протистов до образования пурина. регулирующие сайты. Это говорит о том, что есть некоторое избирательное преимущество самой антенны и что животные развили новые функции для GLUD1 за счет добавления аллостерическая регуляция.[7]
GLUD1 может образовывать длинные волокна за счет объединения гексамеров из конца в конец. Полимеризация не связана с каталитической активностью, но, вероятно, играет важную роль, такую как образование мультиферментных комплексов.
GLUD1 имеет два сайта связывания коферментов: один в NAD-BD, который способен связывать эфир NAD + или NADP.+ и непосредственно участвует в каталитическом процессе, а второй, который выполняет регуляторную функцию, находится непосредственно под стержневой спиралью и может связывать АДФ, НАД+, или НАДН, но плохо связывает НАДФН.[8]
Функция
GLUD1 катализирует окислительное дезаминирование Glu до 2-оксоглутарата и свободного NH4+ используя либо NAD+ или НАДФ+ как сопутствующий фактор. Реакция происходит с переносом иона гидрида от Cα Glu к NAD (P).+, тем самым образуя 2-иминоглутарат, который гидролизуется до 2-оксоглутарата и NH4+. Равновесие реакции при стандартных условиях в значительной степени способствует образованию Glu по сравнению с NH.4+ (Go '~ 30 кДж · моль-1) образование. По этой причине считалось, что этот фермент играет важную роль в детоксикации аммиака, поскольку, поскольку высокий [NH4+] токсичны, это положение равновесия было бы физиологически важным; это поможет поддерживать низкий уровень [NH4+]. Однако у людей с определенной формой гипераммониемия в результате формы гиперинсулинизм, активность фермента увеличивается из-за снижения чувствительности к GTP, негативному регулятору. У этих людей уровень аммиака в крови значительно повышен, чего нельзя было бы ожидать, если бы фермент действительно работал в равновесии.
Взаимодействия
Связующие партнеры
ADP
АДФ связывается позади НАД-БД, прямо под центральной спиралью - вторым сайтом связывания кофермента. Аденозиновый фрагмент связывается в гидрофобном кармане с рибозофосфатными группами, направленными вверх в сторону центральной спирали.
ADP также может связываться со вторым, ингибирующим, NADH-сайтом, но вызывает активацию.
GTP
Связывание GTP антагонизирует Pя и АДФ, но является синергическим с НАДН, связанным в некаталитическом аллостерическом сайте. Большинство контактов между GTP и ферментом осуществляется через трифосфатный фрагмент. Сайт связывания GTP считается «сенсором», который выключает фермент, когда клетка находится в состоянии высокой энергии. GTP связывается на стыке между NAD-BD и антенной.[8][9]
В то время как большинство взаимодействий GLUD1-GTP осуществляется через β- и γ-фосфатные взаимодействия, существуют специфические взаимодействия с E346 и K343, которые отдают предпочтение гуанозину по сравнению с аденозином.
В открытой конформации сайт связывания GTP искажен, так что он больше не может связывать GTP.[6]
Регулирование
Когда GLUD1 сильно насыщен лигандами (субстратами) активного центра, в активном центре образуется ингибирующий абортивный комплекс: NAD (P) H.Glu в реакции окислительного дезаминирования при высоком pH и NAD (P).+.2-оксоглутарат в реакции восстановительного аминирования при низком pH. GLUD1 принимает конфигурацию своего базального состояния в отсутствие аллостерических эффекторов, независимо от того, функционируют ли аллостерические сайты. Аллостерические регуляторы GLUD1 - ADP, GTP, Leu, NAD.+ и НАДН - проявляют свои эффекты, изменяя энергию, необходимую для открытия и закрытия каталитической щели во время ферментативного обмена, другими словами, дестабилизируя или стабилизируя, соответственно, абортивные комплексы. Активаторы не нужны для каталитической функции GLUD1, так как он активен в отсутствие этих соединений (базальное состояние). Было высказано предположение, что GLUD1 принимает в своем базальном состоянии конфигурацию (открытая каталитическая щель), которая допускает каталитическую активность независимо от того, являются ли аллостерические сайты функциональными. Регуляция GLUD имеет особое биологическое значение, что подтверждается наблюдениями, показывающими, что регуляторные мутации GLUD1 связаны с клиническими проявлениями у детей.
ADP
АДФ, являющийся одним из двух основных активаторов (НАД+ будучи другим), действует, дестабилизируя дестабилизирующие комплексы и уничтожая негативное сотрудничество. В отсутствие субстратов и со связанным АДФ каталитическая щель находится в открытой конформации, и гексамеры GLUD1 образуют длинные полимеры в кристаллической ячейке с большим количеством взаимодействий, чем обнаруженные в абортивных сложных кристаллах (1NQT ). Это согласуется с тем фактом, что ADP способствует агрегации в растворе. Когда каталитическая щель открывается, R516 превращается в фосфаты АДФ.[8] Раскрытие каталитической щели примерно коррелирует с расстоянием между R516 и фосфатами АДФ. Таким образом, ADP активирует GLUD1, облегчая открытие каталитической щели, что снижает сродство продукта и облегчает высвобождение продукта.[6][10] тем самым позволяя GLUD1 согласовывать некаталитические абортивные комплексы.[9]
Ранее предполагалось, что ингибирование высоким [АДФ] связано с конкуренцией между АДФ и аденозиновым фрагментом кофермента в активном центре1. По крайней мере, известно, что на эффект относительно не влияют ни H507Y, ни R516A.
АТФ
АТФ оказывает комплексное зависящее от концентрации влияние на активность GLUD1:
- Низкое [АТФ] - ингибирование, опосредованное через сайт связывания GTP, поскольку оно устраняется H507Y. Сродство АТФ к сайту GTP, по-видимому, в 1000 раз ниже, чем к GTP, поскольку взаимодействия β- и γ-фосфата являются основными детерминантами связывания на сайте GTP.
- Промежуточное звено [АТФ] - активация, опосредованная эффекторным сайтом АДФ, так как практически полностью устраняется R516A. На этом сайте нуклеотидная группа является главной детерминантой связывания.
- Высокое [АТФ] - ингибирование, опосредованное слабым связыванием в третьем сайте, которое относительно специфично для нуклеотидов аденина. На этот эффект относительно не влияют ни H507Y, ни R516A. Как предполагалось для АДФ, это могло быть связано с конкуренцией между АТФ и аденозиновым фрагментом кофермента в активном центре.[11]
GTP
GTP подавляет оборот ферментов в широком диапазоне условий, увеличивая сродство GLUD1 к продукту реакции, делая скорость высвобождения продукта ограничивающей во всех условиях в присутствии GTP. GTP действует, удерживая каталитическую щель в закрытой конформации, таким образом стабилизируя абортивные комплексы. Эффекты GTP на GLUD1 не локализованы исключительно в субъединице, с которой он связывается, и что антенна играет важную роль в передаче этого ингибирования другим субъединицам.
Лея
Leu активирует GLUD1 независимо от сайта ADP путем связывания в другом месте, возможно, непосредственно внутри каталитической щели. Усиленные ответы пациентов с HI / HA (см. Синдром HI / HA) на стимуляцию Leu высвобождения INS3, которая является результатом их ослабленной чувствительности к ингибированию GTP, подчеркивают физиологическое значение ингибирующего контроля GLUD1.[11]
НАД+
NAD (P) (H) может связываться со вторым сайтом каждой субъединицы. Этот сайт связывает НАД (Н) в ~ 10 раз лучше, чем НАДФ (Н), с восстановленными формами лучше, чем с окисленными. Хотя было высказано предположение, что связывание восстановленного кофермента на этом сайте ингибирует реакцию, в то время как связывание окисленного кофермента вызывает активацию, эффект до сих пор не ясен.
НАДН
НАДН - еще один главный аллостерический ингибитор GLUD1.
Фосфат
Фосфат и другие двухвалентные анионы стабилизируют GLUD1. Недавние структурные исследования показали, что молекулы фосфата связываются с сайтом GTP.[8]
Клиническое значение
Семейный гиперинсулинизм, связанный с мутациями в GLUD1, характеризуется гипогликемией, которая варьируется от тяжелого неонатального начала, трудно поддающегося лечению заболевания, до заболевания, начинающегося в детстве, с легкими симптомами и трудно диагностируемыми. гипогликемия. Неонатальное заболевание проявляется в течение нескольких часов или двух дней после рождения. Заболевание, начавшееся в детстве, проявляется в первые месяцы или годы жизни. В период новорожденности симптомы могут быть неспецифическими, включая судороги, гипотонию, плохое питание и апноэ. В тяжелых случаях концентрации глюкозы в сыворотке крови обычно чрезвычайно низкие и поэтому легко распознаются, тогда как в более легких случаях вариабельная и легкая гипогликемия может затруднить диагностику. Даже в пределах одной семьи проявления болезни могут варьироваться от легких до тяжелых. Лица с аутосомно-рецессивным семейным гиперинсулинизмом, вызванным мутациями в ABCC8 или KCNJ11 (FHI-KATP), имеют тенденцию быть большими для гестационного возраста и обычно проявляются тяжелой рефрактерной гипогликемией в первые 48 часов жизни; Пострадавшие младенцы обычно лишь частично реагируют на диету или лечение (например, терапию диазоксидом) и, следовательно, могут потребовать резекции поджелудочной железы. Лица с аутосомно-доминантным FHI-КАТП обычно подходят для гестационного возраста при рождении, примерно в возрасте одного года (диапазон: от 2 дней до 30 лет) и для ответа на диету и терапию диазоксидом. Сообщалось об исключениях из обоих этих общих положений. FHI-GCK, вызванный мутациями в GCK, может быть намного мягче, чем FHI-KATP; однако у некоторых людей наблюдается тяжелая гипогликемия, не реагирующая на диазоксид. FHI-HADH, вызванный мутациями в HADH, имеет тенденцию быть относительно легким, хотя сообщалось о тяжелых случаях. Люди с FHI-HNF4A, вызванным мутациями в HNF4A, обычно рождаются крупными для гестационного возраста и имеют легкие черты характера, которые реагируют на диазоксид лечение. FHI-UCP2, вызванный мутациями в UCP2, является редкой причиной FH1, чувствительного к диазоксиду. Гипераммониемия / гиперинсулинизм (HA / HI) связан с гипераммониемией от легкой до умеренной и с относительно легкой гипогликемией с поздним началом; большинство, но не все пораженные люди имеют мутации в GLUD1.[12]
Клинические характеристики
FHI характеризуется гипогликемией, которая варьируется от тяжелого неонатального начала, трудно поддающегося лечению заболевания, до заболевания, начинающегося в детстве, с легкими симптомами и трудно диагностируемой гипогликемией. Неонатальное заболевание проявляется в течение нескольких часов или двух дней после рождения. Заболевание, начавшееся в детстве, проявляется в первые месяцы или годы жизни.[13] В период новорожденности симптомы могут быть неспецифическими, включая судороги, гипотонию, плохое питание и апноэ. В тяжелых случаях концентрации глюкозы в сыворотке крови обычно чрезвычайно низкие и поэтому легко распознаются, тогда как в более легких случаях вариабельная и легкая гипогликемия может затруднить диагностику. Даже в пределах одной семьи проявления болезни могут варьироваться от легких до тяжелых.[14]
Диагностика / тестирование
Приблизительно 45% больных имеют мутации либо в ABCC8, который кодирует белок SUR1, либо в KCNJ11, который кодирует белок Kir6.2. В еврейской популяции ашкенази две мутации-основатели ABCC8 ответственны за приблизительно 97% FHI. Другие мутации-основатели ABCC8 присутствуют в финской популяции (p. Val187Asp и p.Asp1506Lys). Каждая мутация в GLUD1 и HNF4A составляет примерно 5% людей с FHI.[15][16] Активирующие мутации в GCK или инактивирующие мутации в HADH встречаются менее чем у 1% людей с FHI. На сегодняшний день мутации в UCP2 описаны только в двух семьях. Приблизительно 40% людей с FHI не имеют идентифицируемой мутации ни в одном из генов, которые, как известно, связаны с FHI.
Управление
При первоначальном диагнозе гипогликемия корректируется внутривенным введением глюкозы для нормализации концентрации глюкозы в плазме и предотвращения повреждения мозга.[17] Долгосрочное лечение включает использование диазоксида, аналогов соматостатина, нифедипина, глюкагона, рекомбинантного IGF-I, глюкокортикоидов, гормона роста человека, диетических вмешательств или комбинаций этих методов лечения.[18] У пациентов, у которых агрессивное медицинское лечение не позволяет поддерживать концентрацию глюкозы в плазме в безопасных пределах или у которых такая терапия не может безопасно поддерживаться в течение долгого времени, рассматривается резекция поджелудочной железы.[19]
использованная литература
- ^ а б c ГРЧ38: Ансамбль выпуск 89: ENSG00000148672 - Ансамбль, Май 2017
- ^ а б c GRCm38: выпуск Ensembl 89: ENSMUSG00000021794 - Ансамбль, Май 2017
- ^ "Справочник человека по PubMed:". Национальный центр биотехнологической информации, Национальная медицинская библиотека США.
- ^ "Ссылка на Mouse PubMed:". Национальный центр биотехнологической информации, Национальная медицинская библиотека США.
- ^ Шашидхаран П., Михаэлидис Т.М., Робакис Н.К., Кресовали А., Папаматеакис Дж., Плайтакис А. (июнь 1994 г.). «Новая человеческая глутаматдегидрогеназа, экспрессируемая в нервных тканях и тканях яичек и кодируемая X-связанным геном без интрона». J. Biol. Chem. 269 (24): 16971–6. PMID 8207021.
- ^ а б c Смит Т.Дж., Шмидт Т., Фанг Дж., Ву Дж., Сюздак Г, Стэнли, Калифорния (май 2002 г.). «Структура апо человеческой глутаматдегидрогеназы детализирует связь субъединиц и аллостерию». J. Mol. Биол. 318 (3): 765–77. Дои:10.1016 / S0022-2836 (02) 00161-4. PMID 12054821.
- ^ Банерджи С., Шмидт Т., Фанг Дж., Стэнли, Калифорния, Смит Т.Дж. (апрель 2003 г.). «Структурные исследования активации АДФ глутаматдегидрогеназы млекопитающих и эволюции регуляции». Биохимия. 42 (12): 3446–56. Дои:10.1021 / bi0206917. PMID 12653548.
- ^ а б c d Смит Т.Дж., Петерсон П.Е., Шмидт Т., Фанг Дж., Стэнли, Калифорния (март 2001 г.). «Структура бычьих глутаматдегидрогеназных комплексов проясняет механизм пуриновой регуляции». J. Mol. Биол. 307 (2): 707–20. Дои:10.1006 / jmbi.2001.4499. PMID 11254391.
- ^ а б Peterson PE, Smith TJ (июль 1999 г.). «Структура бычьей глутаматдегидрогеназы дает представление о механизме аллостерии». Структура. 7 (7): 769–82. Дои:10.1016 / S0969-2126 (99) 80101-4. PMID 10425679.
- ^ Джордж А., Белл Дж. Э. (декабрь 1980 г.). «Влияние аденозин-5'-дифосфата на глутаматдегидрогеназу крупного рогатого скота: модификация диэтилпирокарбоната». Биохимия. 19 (26): 6057–61. Дои:10.1021 / bi00567a017. PMID 7470450.
- ^ а б Фанг, Дж; Hsu, BY; MacMullen, CM; Понц, М; Смит, Т.Дж.; Стэнли, Калифорния (2002). «Экспрессия, очистка и характеристика аллостерических регуляторных мутаций GLUD1». Biochem. J. 363 (Пт 1): 81–7. Дои:10.1042/0264-6021:3630081. ЧВК 1222454. PMID 11903050.
- ^ «Энтрез Ген: глутаматдегидрогеназа 1».
- ^ Вон Дж.Г., Ценг Х.С., Ян А.Х., Тан К.Т., Яп Т.С., Ли СН, Линь HD, Буркус Н., Питтенгер Джи, Виник А. (ноябрь 2006 г.). «Клинические особенности и морфологическая характеристика 10 пациентов с инсулиномным панкреатогенным гипогликемическим синдромом (NIPHS)». Клиническая эндокринология. 65 (5): 566–78. Дои:10.1111 / j.1365-2265.2006.02629.x. PMID 17054456. S2CID 19076202.
- ^ Пинни С.Е., МакМаллен С., Беккер С., Лин Ю.В., Ханна С., Торнтон П., Гангули А., Шинг С.Л., Стэнли, Калифорния (август 2008 г.). «Клиническая характеристика и биохимические механизмы врожденного гиперинсулинизма, связанного с мутациями доминантного канала КАТФ». Журнал клинических исследований. 118 (8): 2877–86. Дои:10.1172 / JCI35414. ЧВК 2441858. PMID 18596924.
- ^ Глейзер Б., Блех И., Кракиновский Ю., Экштейн Дж., Гиллис Д., Мазор-Аронович К., Ландау Г., Абелиович Д. (октябрь 2011 г.). «Частота аллеля мутации ABCC8 в еврейской популяции ашкенази и риск очаговой гиперинсулинемической гипогликемии». Генетика в медицине. 13 (10): 891–4. Дои:10.1097 / GIM.0b013e31821fea33. PMID 21716120. S2CID 11352891.
- ^ Хойлунд К., Хансен Т., Лайер М., Хенриксен Дж. Э., Левин К., Линдхольм Дж., Педерсен О., Бек-Нильсен Х. (июнь 2004 г.). «Новый синдром аутосомно-доминантной гиперинсулинемической гипогликемии, связанный с мутацией в гене рецептора человеческого инсулина». Сахарный диабет. 53 (6): 1592–8. Дои:10.2337 / диабет.53.6.1592. PMID 15161766.
- ^ Мазор-Аронович К., Ландау Х., Гиллис Д. (март 2009 г.). «Хирургическое и нехирургическое лечение врожденного гиперинсулинизма». Обзоры детской эндокринологии. 6 (3): 424–30. PMID 19396028.
- ^ Мазор-Аронович К., Гиллис Д., Лобель Д., Хирш Х. Дж., Пинхас-Хамиэль О., Модан-Мозес Д., Глейзер Б., Ландау Х (октябрь 2007 г.). «Отдаленные результаты нервного развития при консервативном лечении врожденного гиперинсулинизма». Европейский журнал эндокринологии. 157 (4): 491–7. Дои:10.1530 / EJE-07-0445. PMID 17893264.
- ^ Стэнли К.А., Торнтон П.С., Гангули А., МакМаллен С., Андервуд П., Бхатиа П., Стейнкраусс Л., Ваннер Л., Кай Р., Ручелли Е., Сучи М., Адзик Н.С. (январь 2004 г.). «Предоперационная оценка младенцев с очаговым или диффузным врожденным гиперинсулинизмом с помощью внутривенных тестов на острый инсулиновый ответ и селективной стимуляции кальциевой артерии поджелудочной железы». Журнал клинической эндокринологии и метаболизма. 89 (1): 288–96. Дои:10.1210 / jc.2003-030965. PMID 14715863.
внешняя ссылка
- GeneReviews / NCBI / NIH / UW запись о семейном гиперинсулинизме
- Глутамат + дегидрогеназа в Национальной медицинской библиотеке США Рубрики медицинской тематики (MeSH)
- Обзор всей структурной информации, доступной в PDB за UniProt: P00367 (Глутаматдегидрогеназа 1, митохондриальная) на PDBe-KB.
Галерея PDB | |
|---|---|
|






Эта статья включает текст из Национальная медицинская библиотека США, который находится в всеобщее достояние.