ATP7A - ATP7A





ATP7A, также известный как Протеин Менкеса (MNK), является медь транспортирующим АТФаза P-типа который использует энергию, возникающую из Гидролиз АТФ для транспортировки Cu (I) через клеточные мембраны. Белок ATP7A представляет собой трансмембранный белок и экспрессируется в кишечнике и во всех тканях, кроме печени. В кишечнике ATP7A регулирует всасывание Cu (I) в организме человека, транспортируя Cu (I) из тонкого кишечника в кровь. В других тканях ATP7A перемещается между аппарат Гольджи и клеточная мембрана для поддержания надлежащих концентраций Cu (I) (поскольку в клетке нет свободной Cu (I), ионы Cu (I) все прочно связаны) в клетке и обеспечивает определенные ферменты Cu (I) (например, пептидил-α-монооксигеназа, тирозиназа, и лизилоксидаза ). Х-сцепленное наследственное летальное генетическое заболевание ATP7A генные причины Болезнь Менкеса, дефицит меди, приводящий к смерти в раннем детстве.[5]
Ген
В ATP7A ген расположен на длинном (q) плече Х хромосома в полосе Xq21.1. Кодируемый белок ATP7A состоит из 1500 аминокислот.[6] Мутации / добавления / делеции этого гена часто вызывают дефицит меди, что приводит к прогрессирующей нейродегенерации и смерти у детей.[7]
Структура
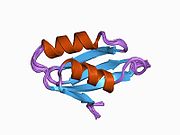
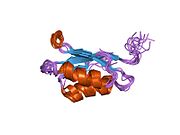
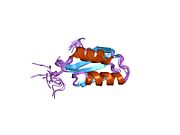
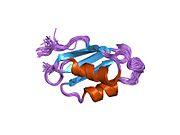
ATP7A - это трансмембранный белок причем оба N- и C-конца ориентированы в сторону цитозоля (см. рисунок). Он очень гомологичен белку ATP7B. ATP7A содержит три основных функциональных домена:[8][9][10][11]
- 8 трансмембранные сегменты которые образуют канал и позволяют Cu (I) проходить через мембрану;
- АТФ-связывающий домен;
- Большой N-концевой цитозольный домен, который содержит шесть повторяющихся сайтов связывания Cu (I), каждый из которых содержит мотив GMTCXXC.
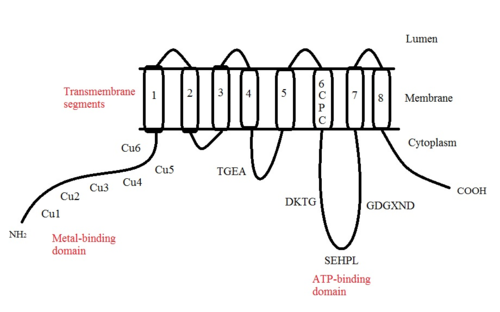
Многие мотивы в структуре ATP7A консервативны:[10]
- Мотив TGEA расположен в петле на цитозольной стороне между трансмембранными сегментами 4 и 5 и участвует в передаче энергии.
- Мотив CPC, расположенный в трансмембранном сегменте 6, является общим для всех АТФаз, транспортирующих тяжелые металлы.
Между трансмембранными сегментами 6 и 7 находится большая цитоплазматическая петля, в которой расположены три мотива: DKTG, SEHPL и GDGXND.
- Мотив DKTG необходим для правильного функционирования АТФазы. В аспарагиновая кислота (D) остаток фосфорилированный во время транспортных циклов.
- Мотив SEHPL существует только в АТФазах P-типа, транспортирующих тяжелые металлы. Без гистидин (H) остаток ATP7A может не функционировать должным образом.
- Считается, что мотив GDGXND рядом с трансмембранным сегментом 7 содержит в основном α-спирали и служит структурной опорой.
Шесть сайтов связывания Cu (I) на N-конце связывают по одному Cu (I) каждый. Этот сайт связывания не специфичен для Cu (I) и может связывать различные ионы переходных металлов. Cd (II), Au (III) и Hg (II) связываются с сайтом связывания более прочно, чем Zn (II), тогда как Mn (II) и Ni (II) имеют более низкое сродство по сравнению с Zn (II). В случае Cu (I) возможный кооперативная привязка механизм наблюдается. Когда концентрация Cu (I) низкая, Cu (I) имеет более низкое сродство к ATP7A по сравнению с Zn (II); по мере увеличения концентрации Cu (I) наблюдается резкое возрастание сродства Cu (I) к белку.[10]
Конформационное изменение
Два цистеин (C) остатки в каждом сайте связывания Cu (I) координированы с Cu (I) с углом S-Cu (I) -S от 120 до 180 ° и расстоянием Cu-S 2,16 Å. Экспериментальные результаты гомологичного белка ATP7B предполагают, что участвуют восстанавливающие реагенты, и при связывании Cu (I) дисульфидная связь между остатками цистеина нарушается, когда цистеин начинает связываться с Cu (I), что приводит к серии конформационных изменений на N-конце белка и, возможно, активирует активность по транспортировке Cu (I) других цитозольных петель.[10]
Из шести сайтов связывания меди (I) двух считается достаточно для функции транспорта Cu (I). Причина, по которой существует шесть сайтов связывания, до конца не выяснена. Однако некоторые ученые предположили, что другие четыре участка могут служить детектором концентрации Cu (I).[8]
Транспортный механизм
ATP7A принадлежит к семейству транспортеров, называемых АТФазы P-типа, которые катализируют авто-фосфорилирование сохраненного ключа аспарагиновая кислота (D) остаток в ферменте. Первым шагом является связывание АТФ с АТФ-связывающим доменом и связывание Cu (I) с трансмембранной областью. Затем ATP7A фосфорилируется по ключевому аспарагиновая кислота (D) остаток в высококонсервативном мотиве DKTG, сопровождающийся высвобождением Cu (I). Последующий дефосфорилирование промежуточного продукта завершает каталитический цикл. В каждом цикле ATP7A преобразуется по крайней мере между двумя различными конформациями, E1 и E2. В состоянии E1 Cu (I) прочно связан с сайтами связывания на цитоплазматической стороне; в состоянии E2 сродство ATP7A к Cu (I) снижается, и Cu (I) высвобождается на внеклеточной стороне.[12]
Функция
ATP7A важен для регуляции меди Cu (I) у млекопитающих.[9] Этот белок содержится в большинстве тканей, но не экспрессируется в печени.[10] В тонком кишечнике белок ATP7A помогает контролировать всасывание Cu (I) из пищи. После поглощения ионов Cu (I) в энтероциты, ATP7A требуется для передачи их через базолатеральная мембрана в обращение.[8]
В других органах и тканях белок ATP7A выполняет двойную роль и перемещается между двумя участками внутри клетки. Белок обычно находится в клеточной структуре, называемой аппарат Гольджи, который модифицирует и транспортирует вновь продуцируемые ферменты и другие белки. Здесь ATP7A поставляет Cu (I) определенным ферментам (например, пептидил-α-монооксигеназа, тирозиназа, и лизилоксидаза[8]), которые имеют решающее значение для структур и функций мозга, костей, кожи, волос, соединительной ткани и нервной системы. Однако, если уровни Cu (I) в клеточной среде повышены, ATP7A перемещается к клеточной мембране и удаляет избыток Cu (I) из клетки.[7][9]
Функции ATP7A в некоторых тканях человеческого тела следующие:[9]
| Ткань | Место расположения | Функция |
|---|---|---|
| Почка | Выражено в эпителиальные клетки проксимального и дистального почечные канальцы | Удаляет избыток Cu (I) для поддержания уровня Cu (I) в почках |
| Паренхима | в цитотрофобласт, синцитиотрофобласт и сосуды плода эндотелиальный клетки | Доставляет Cu (I) плацентарным купроэнзимам и транспортирует Cu (I) в кровообращение плода |
| Центральная нервная система | Различные локации | Распределяет Cu (I) в различных отделах центральной нервной системы. |
Взаимодействия
Было показано, что ATP7A взаимодействует с ATOX1 и GLRX. Антиоксидант 1 медный шаперон (ATOX1) необходим для поддержания гомеостаза меди Cu (I) в клетке. Он может связывать и транспортировать цитозольный Cu (I) к ATP7A в транс-сети Гольджи. Глутаредоксин-1 (GRX1) также важен для функции ATP7A. Он способствует связыванию Cu (I) для последующего транспорта, катализируя восстановление дисульфидных мостиков. Он также может катализировать де-глутатионилирование реакция остатков C (цистеина) в шести Cu (I) -связывающих мотивах GMTCXXC.[9]
Клиническое значение
Болезнь Менкеса это вызвано мутации в гене ATP7A.[13] Исследователи идентифицировали различные мутации ATP7A, которые вызывают болезнь Менкеса и синдром затылочного рога (OHS), более легкая форма болезни Менкеса. Многие из этих мутаций удаляют часть гена и, как предполагается, продуцируют укороченный белок ATP7A, который не может транспортировать Cu (I). Другие мутации вставляют дополнительные пары оснований ДНК или используют неправильные пары оснований, что приводит к белкам ATP7A, которые не функционируют должным образом.[6]
Измененные белки, возникающие в результате мутаций ATP7A, ухудшают абсорбцию меди из пищи, не могут поставлять медь определенным ферментам или застревают в клеточной мембране, не имея возможности перемещаться назад и вперед от Гольджи. В результате нарушенной активности белка ATP7A медь плохо распределяется по клеткам организма. Медь накапливается в некоторых тканях, таких как тонкий кишечник и почки, в то время как в мозге и других тканях ее уровень необычно низкий.[7][8] Уменьшение поступления меди может снизить активность множества медьсодержащих ферментов, которые необходимы для структуры и функции костей, кожи, волос, кровеносных сосудов и нервной системы.[7][9] Медь также имеет решающее значение для распространения прион белки, а мыши с мутациями в Atp7a имеют отсроченное начало прионной болезни. [14] Доступен исчерпывающий ресурс клинически аннотированных генетических вариантов гена ATP7A.[15] подтверждая Американский колледж медицинской генетики и геномики рекомендации по интерпретации вариантов последовательностей.
Торможение
Было показано, что ингибитор протонной помпы, омепразол, блокирует ATP7A в дополнение к его более известной роли блокирования ATP4A.
Рекомендации
- ^ а б c ГРЧ38: Ансамбль выпуск 89: ENSG00000165240 - Ансамбль, Май 2017
- ^ а б c GRCm38: выпуск Ensembl 89: ENSMUSG00000033792 - Ансамбль, Май 2017
- ^ "Справочник человека по PubMed:". Национальный центр биотехнологической информации, Национальная медицинская библиотека США.
- ^ "Ссылка на Mouse PubMed:". Национальный центр биотехнологической информации, Национальная медицинская библиотека США.
- ^ Тюмер З., Мёллер Л. Б., Хорн Н. (1999). Спектр мутаций ATP7A, гена, дефектного при болезни Менкеса. Adv. Exp. Med. Биол. Успехи экспериментальной медицины и биологии. 448. С. 83–95. Дои:10.1007/978-1-4615-4859-1_7. ISBN 978-1-4613-7204-2. PMID 10079817.
- ^ а б Кодама Х., Мурата Й. (август 1999 г.). «Молекулярная генетика и патофизиология болезни Менкеса». Международная педиатрия. 41 (4): 430–5. Дои:10.1046 / j.1442-200x.1999.01091.x. PMID 10453200.
- ^ а б c d е Луценко С., Гупта А., Беркхед Дж. Л., Зузель В. (август 2008 г.). «Многозадачность клетки: двойная роль Cu-АТФаз человека в доставке кофактора и балансе внутриклеточной меди». Архивы биохимии и биофизики. 476 (1): 22–32. Дои:10.1016 / j.abb.2008.05.005. ЧВК 2556376. PMID 18534184.
- ^ а б c d е Бертини I, Грей Х, Штифель Э, Валентайн Дж (2006). Биологическая неорганическая химия: структура и реакционная способность. Саусалито, Калифорния: Университетские научные книги. ISBN 978-1-891389-43-6.
- ^ Инеси Г., Пиланкатта Р., Тадини-Буонинсеньи Ф. (октябрь 2014 г.). «Биохимическая характеристика медных АТФаз Р-типа». Биохимический журнал. 463 (2): 167–76. Дои:10.1042 / BJ20140741. ЧВК 4179477. PMID 25242165.
- ^ Banci L, Bertini I, Cantini F, Ciofi-Baffoni S (август 2010 г.). «Распределение меди в клетках: подход механистической системной биологии». Клеточные и молекулярные науки о жизни. 67 (15): 2563–89. Дои:10.1007 / s00018-010-0330-х. PMID 20333435.
- ^ Hordyjewska A, Popiołek Ł, Kocot J (август 2014 г.). «Многоликая медь в медицине и лечении». Биометаллы. 27 (4): 611–21. Дои:10.1007 / s10534-014-9736-5. ЧВК 4113679. PMID 24748564.
- ^ Сиггс О.М., Cruite JT, Du X, Rutschmann S, Masliah E, Beutler B, Oldstone MB (август 2012 г.). «Нарушение гомеостаза меди из-за мутации Atp7a задерживает начало прионной болезни». Proc. Natl. Акад. Sci. СОЕДИНЕННЫЕ ШТАТЫ АМЕРИКИ. 109 (34): 13733–8. Дои:10.1073 / pnas.1211499109. ЧВК 3427069. PMID 22869751.
- ^ «ATP7Agen - всеобъемлющий ресурс для клинически аннотированных вариантов гена ATP7A». clingen.igib.res.in. Получено 2020-07-06.
дальнейшее чтение
- Барнс Н, Цивковский Р, Цивковская Н, Луценко С (2005). «Переносящие медь АТФазы, белки болезни Менкеса и Вильсона играют разные роли во взрослом и развивающемся мозжечке». J Biol Chem. 280 (10): 9640–5. Дои:10.1074 / jbc.M413840200. PMID 15634671.
- Гриноу М., Пазе Л., Воскобойник И., Петрис М.Дж., О'Брайен А.В., Камакарис Дж. (2004). «Сигналы, регулирующие перенос Menkes (MNK; ATP7A) медь-транслокационной АТФазы P-типа в поляризованных клетках MDCK». Am J Physiol Cell Physiol. 287 (5): C1463–71. Дои:10.1152 / ajpcell.00179.2004. PMID 15269005.
- Мёллер Л. Б., Тюмер З., Лунд С., Петерсен С., Коул Т., Хануш Р., Зайдель Дж., Йенсен Л. Р., Хорн Н. (2000). «Подобные мутации сайта сплайсинга гена ATP7A приводят к различным фенотипам: классической болезни Менкеса или синдрому затылочного рога». Am J Hum Genet. 66 (4): 1211–20. Дои:10.1086/302857. ЧВК 1288188. PMID 10739752.
- Воскобойник И., Камакарис Дж. (2002). «Менкес АТФаза Р-типа, передающая медь (ATP7A): биохимические и биологические свойства клетки, а также роль в болезни Менкеса». J Bioenerg Biomembr. 34 (5): 363–71. Дои:10.1023 / А: 1021250003104. PMID 12539963.
- Харрис Э.Д., Редди М.С., Цянь Й., Тиффани-Кастильони Э., Маджумдар С., Нельсон Дж. (1999). Множественные формы Cu-АТФазы Менкеса. Adv. Exp. Med. Биол. Успехи экспериментальной медицины и биологии. 448. С. 39–51. Дои:10.1007/978-1-4615-4859-1_4. ISBN 978-1-4613-7204-2. PMID 10079814.
- Кокс DW, Мур SD (2003). «Медь-транспортирующие АТФазы Р-типа и болезни человека». J. Bioenerg. Биомер. 34 (5): 333–8. Дои:10.1023 / А: 1021293818125. PMID 12539960.
- Воскобойник I, Камакарис Дж. (2003). «Менкес АТФаза Р-типа, передающая медь (ATP7A): биохимические и биологические свойства клетки, а также роль в болезни Менкеса». J. Bioenerg. Биомер. 34 (5): 363–71. Дои:10.1023 / А: 1021250003104. PMID 12539963.
- La Fontaine S, Mercer JF (2007). «Торговля медь-АТФазы, ATP7A и ATP7B: роль в гомеостазе меди». Arch. Biochem. Биофизы. 463 (2): 149–67. Дои:10.1016 / j.abb.2007.04.021. PMID 17531189.
- Луценко С., ЛеШейн Е.С., Шинде У (2007). «Биохимические основы регуляции медьтранспортных АТФаз человека». Arch. Biochem. Биофизы. 463 (2): 134–48. Дои:10.1016 / j.abb.2007.04.013. ЧВК 2025638. PMID 17562324.
- Диерик Х.А., Амброзини Л., Спенсер Дж., Гловер Т.В., Мерсер Дж. Ф. (1996). «Молекулярная структура гена болезни Менкеса (ATP7A)». Геномика. 28 (3): 462–9. Дои:10.1006 / geno.1995.1175. PMID 7490081.
- Тюмер З., Вурал Б., Тённесен Т., Челли Дж., Монако А. П., Хорн Н. (1995). «Характеристика структуры экзона гена болезни Менкеса с использованием векторной ПЦР». Геномика. 26 (3): 437–42. Дои:10.1016 / 0888-7543 (95) 80160-Н. PMID 7607665.
- Калер С.Г., Галло Л.К., Гордый В.К., Перси А.К., Марк Й., Сигал Н.А., Гольдштейн Д.С., Холмс С.С., Гал В.А. (1995). «Синдром затылочного рога и умеренный фенотип Менкеса, связанный с мутациями сайта сплайсинга в локусе MNK». Nat. Genet. 8 (2): 195–202. Дои:10.1038 / ng1094-195. PMID 7842019.
- Das S, Levinson B, Whitney S, Vulpe C, Packman S, Gitschier J (1994). «Различные мутации у пациентов с болезнью Менкеса часто приводят к пропуску экзонов». Являюсь. J. Hum. Genet. 55 (5): 883–9. ЧВК 1918324. PMID 7977350.
- Chelly J, Tümer Z, Tønnesen T., Petterson A, Ishikawa-Brush Y, Tommerup N, Horn N, Monaco AP (1993). «Выделение гена-кандидата на болезнь Менкеса, который кодирует потенциальный белок, связывающий тяжелые металлы». Nat. Genet. 3 (1): 14–9. Дои:10.1038 / ng0193-14. PMID 8490646.
- Мерсер Дж. Ф., Ливингстон Дж., Холл В, Пэйнтер Дж. А., Беги С., Чандрасекхараппа С., Локхарт П., Граймс А., Бхаве М., Симиениак Д. (1993). «Выделение частичного гена-кандидата на болезнь Менкеса путем позиционного клонирования». Nat. Genet. 3 (1): 20–5. Дои:10.1038 / ng0193-20. PMID 8490647.
- Вулпе С., Левинсон Б., Уитни С., Пакман С., Гитшиер Дж. (1993). «Выделение гена-кандидата на болезнь Менкеса и доказательства того, что он кодирует АТФазу, транспортирующую медь». Nat. Genet. 3 (1): 7–13. Дои:10.1038 / ng0193-7. PMID 8490659.
- Левинсон Б., Конант Р., Шнур Р., Дас С., Пакман С., Гитшиер Дж. (1997). «Повторяющийся элемент в регуляторной области гена MNK и его делеция у пациента с синдромом затылочного рога». Гм. Мол. Genet. 5 (11): 1737–42. Дои:10.1093 / hmg / 5.11.1737. PMID 8923001.
- Yamaguchi Y, Heiny ME, Suzuki M, Gitlin JD (1997). «Биохимическая характеристика и внутриклеточная локализация белка болезни Менкеса». Proc. Natl. Акад. Sci. СОЕДИНЕННЫЕ ШТАТЫ АМЕРИКИ. 93 (24): 14030–5. Дои:10.1073 / пнас.93.24.14030. ЧВК 19489. PMID 8943055.
- Петрис М.Дж., Мерсер Дж.Ф., Калвенор Дж. Г., Локхарт П., Глисон П.А., Камакарис Дж. (1997). «Регулируемый лигандом транспорт насоса оттока АТФазы меди P-типа Менкеса из аппарата Гольджи к плазматической мембране: новый механизм регулируемого трафика». EMBO J. 15 (22): 6084–95. Дои:10.1002 / j.1460-2075.1996.tb00997.x. ЧВК 452430. PMID 8947031.
- Tümer Z, Lund C, Tolshave J, Vural B, Tønnesen T, Horn N (1997). «Выявление точечных мутаций у 41 неродственного пациента с болезнью Менкеса». Являюсь. J. Hum. Genet. 60 (1): 63–71. ЧВК 1712537. PMID 8981948.
- Диерик Х.А., Адам А.Н., Эскара-Уилке Дж.Ф., Гловер Т.В. (1997). «Иммуноцитохимическая локализация белка транспорта меди Менкеса (ATP7A) в сети транс-Гольджи». Гм. Мол. Genet. 6 (3): 409–16. Дои:10,1093 / чмг / 6.3.409. PMID 9147644.
- Ронсе Н., Мойзард М.П., Робб Л., Тутен А., Виллар Л., Морейн С. (1997). «Переход C2055T в экзоне 8 гена ATP7A связан с пропуском экзона в семействе синдрома затылочного рога». Являюсь. J. Hum. Genet. 61 (1): 233–8. Дои:10.1016 / S0002-9297 (07) 64297-9. ЧВК 1715861. PMID 9246006.
- Gitschier J, Moffat B, Reilly D, Wood WI, Fairbrother WJ (1998). «Структура раствора четвертого металлсвязывающего домена из АТФазы Менкеса, транспортирующей медь». Nat. Struct. Биол. 5 (1): 47–54. Дои:10.1038 / nsb0198-47. PMID 9437429.
внешняя ссылка
- ATP7A + белок, + человеческий в Национальной медицинской библиотеке США Рубрики медицинской тематики (MeSH)
- Статья GeneReviews / NCBI / NIH / UW о нарушениях транспорта меди, связанных с ATP7A, включает: болезнь Менкеса, синдром затылочного рога, дистальную моторную нейропатию, связанную с ATP7A
- Записи OMIM о нарушениях транспорта меди, связанных с ATP7A
- GeneCard
- Человек ATP7A расположение генома и ATP7A страница сведений о генах в Браузер генома UCSC.
PDB галерея | |
|---|---|
|