Валлеровское вырождение - Wallerian degeneration
| Травма нерва | |
|---|---|
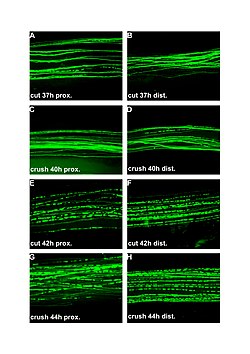 | |
| Флуоресцентные микрофотографии (100x) валлеровской дегенерации перерезанных и раздавленных периферических нервов. Левая колонка проксимальнее травмы, правая - дистальнее. A и B: 37 часов после резки. C и D: 40 часов после увлечения. E и F: 42 часа после резки. G и H: 44 часа после увлечения. | |
| Специальность | Неврология |
Валлеровское вырождение это активный процесс вырождения, который возникает, когда нервное волокно разрезан или раздавлен, а часть аксон дистальнее травмы (т.е. дальше от нейрон клетка тела) дегенерирует.[1] Связанный с этим процесс отмирания или ретроградной дегенерации, известный как «Валлеровская дегенерация», происходит при многих нейродегенеративных заболеваниях, особенно тех, где аксональный транспорт ослаблен, например ALS и Болезнь Альцгеймера.[2] Первичная культура исследования показывают, что неспособность доставить достаточное количество необходимого аксонального белка NMNAT2 является ключевым исходным событием.[3][4]
Валлеровская дегенерация возникает после повреждения аксонов как в периферическая нервная система (ПНС) и Центральная нервная система (ЦНС). Это происходит в отделе аксона дистальнее места повреждения и обычно начинается в течение 24–36 часов после поражения. До дегенерации дистальный отдел аксона имеет тенденцию оставаться электрически возбудимым. После травмы аксональный скелет распадается, и аксональная мембрана разрывается. Дегенерация аксонов сопровождается деградацией миелиновой оболочки и проникновение макрофаги. Макрофаги, сопровождаемые Шванновские клетки, служат для очистки мусора от перерождения.[5][6]
Шванновские клетки реагируют на потерю аксонов экструзией их миелиновых оболочек, подавлением генов миелина, дедифференцировкой и пролиферацией. В конце концов они выстраиваются в трубочки (ленты Бюнгнера) и экспрессируют поверхностные молекулы, направляющие регенерирующие волокна.[7] В течение 4 дней после травмы дистальный конец части нервного волокна, проксимальной к поражению, посылает отростки к этим трубкам, и эти отростки привлекаются факторами роста, продуцируемыми шванновскими клетками в трубках. Если росток достигает трубки, он прорастает в нее и продвигается примерно на 1 мм в день, в конечном итоге достигая и повторно иннервируя ткань-мишень. Если ростки не могут добраться до трубки, например, из-за слишком широкой щели или образования рубцовой ткани, хирургическое вмешательство может помочь направить ростки в трубки. Регенерация ПНС эффективна, с почти полным восстановлением в случае поражений, которые возникают вблизи дистального окончания нервного окончания. Однако выздоровление практически не наблюдается. спинной мозг. Одно важное отличие состоит в том, что в ЦНС, включая спинной мозг, миелиновые оболочки производятся олигодендроциты а не шванновскими клетками.
История
Валлеровское вырождение названо в честь Август Волни Уоллер. Уоллер экспериментировал лягушки в 1850 г., отрезав их языкоглоточный и подъязычный нервы. Затем он осмотрел дистальные нервы от места повреждения, которые были отделены от своих клеточных тел в стволе мозга.[5] Валлер описал распад миелина, который он назвал «мозговым веществом», на отдельные частицы различного размера. Дегенерирующие аксоны образовывали капли, которые можно было окрашивать, что позволяло изучать ход отдельных нервных волокон.
Дегенерация аксонов
Хотя большинство реакций на травмы включают кальций передача сигналов притока, чтобы способствовать повторному закрытию отрубленных частей, повреждения аксонов первоначально приводят к острой дегенерации аксонов (AAD), которая представляет собой быстрое разделение проксимальный (часть ближе к телу клетки) и дистальные концы в течение 30 минут после травмы.[8] После разделения на обоих концах формируются дистрофические луковичные структуры, а перерезанные мембраны герметизируются.[9] В дистальном сегменте происходит короткая латентная фаза, в течение которой он остается электрически возбудимым и структурно неповрежденным.[10] Дегенерация сопровождается набуханием аксолемма, и в конечном итоге образование бусин аксональные сфероиды. Этот процесс занимает около 24 часов в ПНС и дольше в ЦНС. Сигнальные пути, ведущие к дегенерации аксолеммы, в настоящее время плохо изучены. Однако исследования показали, что этот процесс AAD не зависит от кальция.[11]
Гранулярный распад аксонального цитоскелета и внутреннего органеллы возникает после деградации аксолеммы. Ранние изменения включают накопление митохондрии в паранодальные регионы на месте травмы. Эндоплазматический ретикулум распадается, митохондрии набухают и в конечном итоге распадаются. Деполимеризация микротрубочки происходит и вскоре следует за деградацией нейрофиламентов и других компонентов цитоскелета. Распад зависит от Убиквитин и Кальпаин протеазы (вызванные притоком иона кальция), предполагая, что дегенерация аксонов является активным процессом, а не пассивным, как ранее неправильно понималось.[12] Таким образом, аксон подвергается полной фрагментации. Скорость деградации зависит от типа травмы и также медленнее в ЦНС, чем в ПНС. Другим фактором, влияющим на скорость деградации, является диаметр аксона: более крупным аксонам требуется больше времени для деградации цитоскелета и, следовательно, требуется больше времени для дегенерации.
Клиренс миелина
Миелин представляет собой фосфолипидную мембрану, которая оборачивается вокруг аксонов, обеспечивая им изоляцию. Производится Клетки Шванна в ПНС, а олигодендроциты в ЦНС. Очистка от миелина - следующий шаг в дегенерации Валлера после дегенерации аксонов. Очистка от остатков миелина различна для ПНС и ЦНС. ПНС намного быстрее и эффективнее очищает миелиновые остатки по сравнению с ЦНС, и шванновские клетки являются основной причиной этого различия. Другой ключевой аспект - изменение проницаемости гемато-тканевого барьера в двух системах. В ПНС проницаемость увеличивается по всей дистальной культи, но нарушение барьера в ЦНС ограничивается только местом повреждения.[11]
Оформление в ПНС
Реакция шванновских клеток на повреждение аксонов происходит быстро. Период времени до начала дегенерации аксонов. Нейрегулины считаются ответственными за быструю активацию. Они активируют рецепторы ErbB2 в микроворсинках шванновских клеток, что приводит к активации митоген-активированная протеинкиназа (МАПК).[13] Хотя активность MAPK наблюдается, травма зондирование Механизм клеток Шванна еще предстоит полностью понять. «Чувство» сопровождается снижением синтеза липидов миелина и в конечном итоге прекращается в течение 48 часов. Миелиновые оболочки отделяются от аксонов в Резцы Шмидта-Лантермана сначала, а затем быстро разрушаются и укорачиваются, образуя бусинчатые структуры. Шванновские клетки продолжают очищать миелиновый мусор, разрушая собственный миелин, фагоцитоз внеклеточный миелин и привлекает макрофаги к остаткам миелина для дальнейшего фагоцитоза.[11] Однако первые несколько дней макрофаги не притягиваются к региону; следовательно, до этого момента шванновские клетки играют основную роль в очистке миелина.
Было замечено, что клетки Шванна рекрутируют макрофаги путем высвобождения цитокины и хемокины после зондирование аксональной травмы. Привлечение макрофагов помогает улучшить скорость очистки от миелинового мусора. Резидентные макрофаги, присутствующие в нервах, выделяют дополнительные хемокины и цитокины, чтобы привлечь дополнительные макрофаги. Дегенерирующий нерв также производит хемотаксические молекулы макрофагов. Другой источник факторов рекрутирования макрофагов - сыворотка. Задержка рекрутирования макрофагов наблюдалась у мышей с дефицитом В-клеток, лишенных сывороточных антител.[11] Эти сигнальные молекулы вместе вызывают приток макрофагов, пик которого приходится на третью неделю после травмы. В то время как клетки Шванна опосредуют начальную стадию очистки миелинового мусора, макрофаги приходят, чтобы завершить работу. Макрофагам способствуют опсонины, помечающие мусор для удаления. Три основные группы, обнаруженные в сыворотке, включают: дополнять, пентраксины, и антитела. Однако только комплемент помогает при фагоцитозе миелинового мусора.[14]
Муринсон и др. (2005)[15] наблюдали, что немиелинизированные или миелинизированные шванновские клетки при контакте с поврежденным аксоном входят в клеточный цикл, что приводит к пролиферации. Наблюдаемая продолжительность деления шванновских клеток составляла приблизительно 3 дня после повреждения.[16]Возможные источники сигнала пролиферации приписываются рецепторам ErbB2 и рецепторам ErbB3. Эта пролиферация может еще больше повысить скорость очистки миелина и играет важную роль в регенерации аксонов, наблюдаемой в ПНС. Клетки Шванна выделяют факторы роста, которые привлекают новые отростки аксонов, растущие из проксимальной культи после полной дегенерации поврежденной дистальной культи. Это приводит к возможной реиннервации клетки или органа-мишени. Однако реиннервация не обязательно идеальна, так как возможное введение в заблуждение происходит во время реиннервации проксимальных аксонов в клетки-мишени.
Клиренс в ЦНС
По сравнению со шванновскими клетками, олигодендроциты для выживания нуждаются в сигналах аксонов. На стадиях своего развития олигодендроциты, которые не могут установить контакт с аксоном и получить сигналы аксонов, подвергаются апоптоз.[17]
Эксперименты по валлеровской дегенерации показали, что при повреждении олигодендроциты либо подвергаются запрограммированной клеточной гибели, либо переходят в состояние покоя. Следовательно, в отличие от клеток Шванна, олигодендроциты не могут очистить миелиновые оболочки и их остатки. В экспериментах на крысах[18] миелиновые оболочки были обнаружены на срок до 22 месяцев. Следовательно, скорость клиренса миелиновой оболочки в ЦНС очень медленная и, возможно, может быть причиной препятствий в регенерационных возможностях аксонов ЦНС, поскольку отсутствуют факторы роста, привлекающие проксимальные аксоны. Другая особенность, которая в конечном итоге приводит Глиальный шрам формирование. Это еще больше снижает шансы на регенерацию и реиннервацию.
Олигодендроциты не могут привлекать макрофаги для удаления мусора. Поступление макрофагов в участок повреждения ЦНС происходит очень медленно. В отличие от ПНС, Микроглия играют жизненно важную роль в валлеровской дегенерации ЦНС. Однако их рекрутирование происходит медленнее по сравнению с рекрутированием макрофагов в PNS примерно на 3 дня. Более того, микроглия может активироваться, но гипертрофия, и не могут превратиться в полностью фагоцитарные клетки. Те микроглии, которые действительно трансформируются, эффективно очищают от мусора. Дифференцировать фагоцитарную микроглию можно путем тестирования экспрессии Главный комплекс гистосовместимости (MHC) класс I и II при валлеровской дегенерации.[19]Скорость клиренса у микроглии очень низкая по сравнению с макрофагами. Возможный источник вариаций скорости клиренса может включать в себя отсутствие активности опсонина вокруг микроглии и отсутствие повышенной проницаемости в микроглии. гематоэнцефалический барьер. Пониженная проницаемость может еще больше затруднить проникновение макрофагов в место повреждения.[11]
Эти данные позволяют предположить, что задержка валлеровской дегенерации в ЦНС по сравнению с ПНС вызывается не из-за задержки дегенерации аксонов, а, скорее, из-за разницы в скорости клиренса миелина в ЦНС и ПНС.[20]
Регенерация
Регенерация следует за вырождением. Регенерация ПНС происходит быстро, что позволяет возобновлять рост до 1 миллиметра в день.[21] Также могут потребоваться трансплантаты для соответствующей реиннервации. Он поддерживается клетками Шванна за счет высвобождения факторов роста. Регенерация ЦНС происходит намного медленнее и почти отсутствует у большинства позвоночных. Основной причиной этого может быть задержка очистки миелиновых остатков. Миелиновые остатки, присутствующие в ЦНС или ПНС, содержат несколько тормозящих факторов. Продолжительное присутствие миелиновых остатков в ЦНС может препятствовать регенерации.[22]Эксперимент, проведенный на Тритоны, животные, которые обладают способностью к быстрой регенерации аксонов ЦНС, обнаружили, что валлеровская дегенерация повреждения зрительного нерва занимает в среднем от 10 до 14 дней, что также свидетельствует о том, что медленный клиренс тормозит регенерацию.[23]
Шванновские клетки и эндоневральные фибробласты в ПНС
В здоровых нервах, Фактор роста нервов (NGF) производится в очень небольших количествах. Однако при повреждении экспрессия мРНК NGF увеличивается в пять-семь раз в течение 14 дней. Нервные фибробласты и шванновские клетки играют важную роль в повышении экспрессии мРНК NGF.[24] Макрофаги также стимулируют шванновские клетки и фибробласты продуцировать NGF через интерлейкин-1, полученный из макрофагов.[25]Другие нейротрофические молекулы, продуцируемые вместе шванновскими клетками и фибробластами, включают: Нейротрофический фактор головного мозга, Нейротрофический фактор, происходящий из глиальных клеток, Цилиарный нейротрофический фактор, Фактор ингибирования лейкемии, Инсулиноподобный фактор роста, и Фактор роста фибробластов. Вместе эти факторы создают благоприятную среду для роста и регенерации аксонов.[11] Помимо факторов роста, шванновские клетки также обеспечивают структурное руководство для дальнейшего усиления регенерации. Во время фазы пролиферации шванновские клетки начинают формировать линию клеток, называемую Группы Бангнера внутри базальной ламинарной трубки. Было замечено, что аксоны регенерируют в тесной связи с этими клетками.[26]Клетки Шванна активируют выработку молекулы адгезии на клеточной поверхности ниндзюрин, что способствует дальнейшему росту.[27] Эти клеточные линии направляют регенерацию аксона в правильном направлении. Возможным источником ошибки, которая может возникнуть в результате этого, является возможное несовпадение целевых ячеек, как обсуждалось ранее.
Из-за отсутствия таких благоприятных стимулирующих факторов в ЦНС, регенерация в ЦНС задерживается.
Валлеровское вырождение медленное
мышей принадлежащий штамму C57BL /Wlds задержали валлеровское вырождение,[28] и, таким образом, позволяют изучать роли различных типов клеток и лежащих в основе клеточных и молекулярных процессов. Текущее понимание процесса стало возможным благодаря экспериментам на Wlds штамм мышей. Мутация впервые произошла у мышей в Harlan-Olac, лаборатории по производству животных, Соединенное Королевство. В Wlds мутация - это аутосомно-доминантный мутация в хромосоме 4 мыши.[29][30] Мутация гена представляет собой тандемное тройное повторение размером 85 т.п.н., происходящее в природе. Мутировавший регион содержит два связанных гена: никотинамидмононуклеотид аденлилтрансфераза 1 (Nmnat1) и фактор убиквитинирования e4b (Ube4b). Кодировка области линкера 18 аминокислоты также является частью мутации.[6] Защитный эффект WldS белок, как было показано, связан с NAD области NMNAT1+ синтезируя активный сайт.[31]
Хотя созданный белок локализуется в ядре и едва обнаруживается в аксонах, исследования показывают, что его защитный эффект обусловлен его присутствием в аксональном и терминальном компартментах.[32][33] Защита, предоставляемая WldS белок присущ нейронам, а не окружающим опорным клеткам, и только локально защищает аксон, что указывает на то, что внутриклеточный путь ответственен за опосредование валлеровской дегенерации.[34][35]
Эффекты мираS мутация
Мутация не причиняет вреда мыши. Единственный известный эффект заключается в том, что валлеровская дегенерация задерживается в среднем до трех недель после повреждения нерва. Сначала подозревалось, что Wlds Мутация замедляет инфильтрацию макрофагов, но недавние исследования показывают, что мутация защищает аксоны, а не замедляет работу макрофагов.[6] Процесс, с помощью которого достигается защита аксонов, плохо изучен. Однако исследования показывают, что Wlds мутация приводит к увеличению активности NMNAT1, что приводит к увеличению НАД+ синтез.[31] Это, в свою очередь, активирует SIRT1-зависимый процесс в ядре, вызывая изменения в транскрипции генов.[31] НАД+ сам по себе может обеспечить дополнительную защиту аксонов за счет увеличения энергетических ресурсов аксона.[36] Однако более поздние работы вызывают сомнения в том, что либо NMNAT1, либо NAD+ можно заменить на полную длину Wlds ген.[37] Эти авторы продемонстрировали методами in vitro и in vivo, что защитный эффект сверхэкспрессии NMNAT1 или добавления NAD+ не защищает аксоны от дегенерации. Однако более поздние исследования показали, что NMNAT1 является защитным в сочетании с нацеливающим на аксоны пептидом, что позволяет предположить, что ключ к защите, обеспечиваемой WldS представляла собой комбинацию активности NMNAT1 и аксональной локализации, обеспечиваемой N-концевым доменом химерного белка.[38]
Обеспеченная защита аксонов задерживает начало дегенерации Валлера. Следовательно, активация шванновских клеток должна быть отложена, поскольку они не будут обнаруживать сигналы деградации аксонов от рецепторов ErbB2. В экспериментах на Wlds у мутировавших мышей инфильтрация макрофагов значительно задерживалась на срок от шести до восьми дней.[39] Однако, как только деградация аксонов началась, дегенерация протекает нормально, и, в зависимости от нервной системы, деградация следует с вышеописанной скоростью. Возможные последствия этого позднего начала - более слабые регенеративные способности у мышей. Исследования показывают, что регенерация может быть нарушена в WldS мышей, но это, вероятно, результат того, что окружающая среда неблагоприятна для регенерации из-за постоянного существования недогенерированных дистальных волокон, тогда как обычно остатки очищаются, уступая место новому росту.[40]
SARM1
Путь валлеровской дегенерации был дополнительно прояснен открытием, что стерильный альфа- и TIR-мотив, содержащий белок 1 (SARM1), играет центральную роль в пути валлеровской дегенерации. Ген был впервые идентифицирован в Drosophila melanogaster скрининг мутагенеза и последующее нокаутирование его гомолога у мышей показали надежную защиту перерезанных аксонов, сравнимую с защитой WldS.[41][42]
SARM1 катализирует синтез и гидролиз циклическая АДФ-рибоза (cADPR) из НАД+ к АДФ-рибоза.[43] Активация SARM1 локально вызывает быстрый коллапс НАД+ уровни в дистальном отделе поврежденного аксона, который затем подвергается дегенерации.[44] Этот коллапс в НАД+ Позже было показано, что уровни SARM1 TIR домен имеющий собственный НАД+ активность расщепления.[45] Белок SARM1 имеет четыре домена, сигнал митохондриальной локализации, авто-ингибирующую N-концевую область, состоящую из мотивов броненосца / HEAT, два стерильных альфа-мотива, ответственных за мультимеризацию, и C-конец. Toll / рецептор интерлейкина-1 обладающий ферментативной активностью.[45] Активации SARM1 достаточно, чтобы разрушить NAD+ уровни и инициируют путь валлеровской дегенерации.[44]
Активность SARM1 помогает объяснить защитную природу фактора выживания. NMNAT2, поскольку ферменты NMNAT предотвращают опосредованное SARM1 истощение NAD+.[46] Эта взаимосвязь дополнительно подтверждается тем фактом, что мыши, лишенные NMNAT2, которые обычно нежизнеспособны, полностью спасаются путем делеции SARM1, в результате чего активность NMNAT2 располагается выше SARM1.[47] Другие пути передачи сигналов, способствующие дегенерации, такие как путь киназы MAP, были связаны с активацией SARM1. Было показано, что передача сигналов MAPK способствует потере NMNAT2, тем самым способствуя активации SARM1, хотя активация SARM1 также запускает каскад киназ MAP, что указывает на существование некоторой формы петли обратной связи.[48][49] Одно объяснение защитного действия WldS Мутация состоит в том, что область NMNAT1, которая обычно локализована в соме, заменяет лабильный фактор выживания NMNAT2 для предотвращения активации SARM1, когда N-концевой участок Ube4 белка WldS локализует его на аксоне. Тот факт, что улучшенная выживаемость WldS аксонов связано с более медленным оборотом WldS по сравнению с NMNAT2 также помогает объяснить, почему нокаут SARM1 обеспечивает более длительную защиту, поскольку SARM1 будет полностью неактивным независимо от активности ингибитора, тогда как WldS в конечном итоге будет деградировать. Возможные последствия пути SARM1 для здоровья человека могут быть обнаружены на животных моделях, которые демонстрируют травматическое повреждение мозга, как мыши, содержащие Сарм1 удаления в дополнение к WldS показывают снижение аксонов после травмы. [50] Специфические мутации в NMNAT2 связали механизм валлеровской дегенерации с двумя неврологическими заболеваниями.
Смотрите также
Рекомендации
- ^ Травма и валлеровская дегенерация, Калифорнийский университет в Сан-Франциско
- ^ Coleman MP, Freeman MR (1 июня 2010 г.). "Валлеровское вырождение, мир (а) и сущность". Ежегодный обзор нейробиологии. 33 (1): 245–67. Дои:10.1146 / annurev-neuro-060909-153248. ЧВК 5223592. PMID 20345246.
- ^ Джилли Дж., Член парламента Коулмана (январь 2010 г.). «Эндогенный Nmnat2 является важным фактором выживания для поддержания здоровья аксонов». PLOS Биология. 8 (1): e1000300. Дои:10.1371 / journal.pbio.1000300. ЧВК 2811159. PMID 20126265.
- ^ Brazill JM, Li C, Zhu Y, Zhai RG (2017). «NMNAT: Это НАД + синтаза… Это шаперон… Это нейропротектор». Текущее мнение в области генетики и развития. 44: 156–162. Дои:10.1016 / j.gde.2017.03.014. ЧВК 5515290. PMID 28445802.
- ^ а б Уоллер А. (1 января 1850 г.). «Эксперименты на разрезе язычникового и подъязычного нервов лягушки и наблюдения за изменениями в структуре их примитивных волокон».. Философские труды Лондонского королевского общества. 140: 423–429. Дои:10.1098 / рстл.1850.0021. JSTOR 108444.
- ^ а б c Coleman MP, Conforti L, Buckmaster EA, Tarlton A, Ewing RM, Brown MC, Lyon MF, Perry VH (август 1998 г.). "Тандемное тройное повторение 85 kb у мышей с медленной валлеровской дегенерацией (Wlds)". Труды Национальной академии наук Соединенных Штатов Америки. 95 (17): 9985–90. Bibcode:1998PNAS ... 95.9985C. Дои:10.1073 / пнас.95.17.9985. ЧВК 21448. PMID 9707587.
- ^ Stoll G, Müller HW (апрель 1999 г.). «Повреждение нерва, дегенерация аксонов и регенерация нервов: основные выводы». Патология головного мозга. 9 (2): 313–25. Дои:10.1111 / j.1750-3639.1999.tb00229.x. PMID 10219748.
- ^ Kerschensteiner M, Schwab ME, Lichtman JW, Misgeld T (май 2005 г.). «Визуализация дегенерации и регенерации аксонов в травмированном спинном мозге in vivo». Природа Медицина. 11 (5): 572–7. Дои:10,1038 / нм1229. PMID 15821747. S2CID 25287010.
- ^ Эддлман К.С., Баллингер М.Л., Смайерс М.Э., Фишман Н.М., Биттнер Г.Д. (июнь 1998 г.). «Эндоцитозное образование везикул и других мембранных структур, вызванное Са2 + и повреждением аксолеммы». Журнал неврологии. 18 (11): 4029–41. Дои:10.1523 / JNEUROSCI.18-11-04029.1998. ЧВК 6792792. PMID 9592084.
- ^ Ван Дж. Т., Медресс З. А., Баррес Б. А. (январь 2012 г.). «Дегенерация аксонов: молекулярные механизмы пути самоуничтожения». Журнал клеточной биологии. 196 (1): 7–18. Дои:10.1083 / jcb.201108111. ЧВК 3255986. PMID 22232700.
- ^ а б c d е ж Варгас МЭ, Баррес Б.А. (1 июля 2007 г.). «Почему валлеровская дегенерация ЦНС такая медленная?». Ежегодный обзор нейробиологии. 30 (1): 153–79. Дои:10.1146 / annurev.neuro.30.051606.094354. PMID 17506644.
- ^ Циммерман UP, Schlaepfer WW (март 1984 г.). «Множественные формы Са-активированной протеазы из мозга и мышц крысы». Журнал биологической химии. 259 (5): 3210–8. PMID 6321500.
- ^ Гертин А.Д., Чжан Д.П., Мак К.С., Альберта Д.А., Ким Х.А. (март 2005 г.). «Микроанатомия передачи сигналов аксона / глии во время валлеровской дегенерации». Журнал неврологии. 25 (13): 3478–87. Дои:10.1523 / JNEUROSCI.3766-04.2005. ЧВК 6724908. PMID 15800203.
- ^ Дайли А.Т., Веллино А.М., Бентем Л., Сильвер Дж., Клиот М. (сентябрь 1998 г.). «Истощение комплемента снижает инфильтрацию и активацию макрофагов во время дегенерации Валлера и регенерации аксонов». Журнал неврологии. 18 (17): 6713–22. Дои:10.1523 / JNEUROSCI.18-17-06713.1998. ЧВК 6792968. PMID 9712643.
- ^ Муринсон BB, Арчер Д.Р., Ли Й., Гриффин Д.В. (февраль 2005 г.). «Дегенерация миелинизированных эфферентных волокон вызывает митоз в клетках Ремака Шванна неповрежденных афферентов С-волокон». Журнал неврологии. 25 (5): 1179–87. Дои:10.1523 / JNEUROSCI.1372-04.2005. ЧВК 6725954. PMID 15689554.
- ^ Лю Х.М., Ян Л.Х., Ян Й.Дж. (июль 1995 г.). «Свойства шванновских клеток: 3. Экспрессия C-fos, продукция bFGF, фагоцитоз и пролиферация во время валлеровской дегенерации». Журнал невропатологии и экспериментальной неврологии. 54 (4): 487–96. Дои:10.1097/00005072-199507000-00002. PMID 7602323.
- ^ Баррес Б.А., Якобсон М.Д., Шмид Р., Сендтнер М., Рафф М.С. (август 1993 г.). «Зависит ли выживание олигодендроцитов от аксонов?». Текущая биология. 3 (8): 489–97. Дои:10.1016 / 0960-9822 (93) 90039-Q. PMID 15335686. S2CID 39909326.
- ^ Людвин СК (31 мая 1990 г.). «Выживание олигодендроцитов при валлеровской дегенерации». Acta Neuropathologica. 80 (2): 184–91. Дои:10.1007 / BF00308922. PMID 1697140. S2CID 36103242.
- ^ Кошинага М., Уиттемор С.Р. (апрель 1995 г.). «Временная и пространственная активация микроглии в трактах волокон, подвергающихся антероградной и ретроградной дегенерации после поражения спинного мозга». Журнал нейротравмы. 12 (2): 209–22. Дои:10.1089 / neu.1995.12.209. PMID 7629867.
- ^ Джордж Р., Гриффин Дж. В. (октябрь 1994 г.). «Отсроченные ответы макрофагов и клиренс миелина во время Валлеровской дегенерации в центральной нервной системе: модель дорсальной радикулотомии». Экспериментальная неврология. 129 (2): 225–36. Дои:10.1006 / exnr.1994.1164. PMID 7957737. S2CID 40089749.
- ^ Ланди-Экман Л. (2007). Неврология: основы реабилитации (3-е изд.). Сондерс. ISBN 978-1-4160-2578-8.
- ^ He Z, Koprivica V (21 июля 2004 г.). «Сигнальный путь Nogo для блока регенерации». Ежегодный обзор нейробиологии. 27 (1): 341–68. Дои:10.1146 / annurev.neuro.27.070203.144340. PMID 15217336.
- ^ Тернер Дж. Э., Глейз К. А. (март 1977 г.). «Ранние стадии валлеровской дегенерации поврежденного зрительного нерва тритона (Triturus viridescens)». Анатомический рекорд. 187 (3): 291–310. Дои:10.1002 / ар.1091870303. PMID 851236.
- ^ Heumann R, Korsching S, Bandtlow C, Thoenen H (июнь 1987 г.). «Изменения синтеза фактора роста нервов в ненейрональных клетках в ответ на перерезку седалищного нерва» (PDF). Журнал клеточной биологии. 104 (6): 1623–31. Дои:10.1083 / jcb.104.6.1623. ЧВК 2114490. PMID 3034917.
- ^ Линдхольм Д., Хойманн Р., Хенгерер Б., Тёнен Х. (ноябрь 1988 г.). «Интерлейкин 1 увеличивает стабильность и транскрипцию мРНК, кодирующей фактор роста нервов, в культивируемых фибробластах крыс». Журнал биологической химии. 263 (31): 16348–51. PMID 3263368.
- ^ Томас П.К., король Р.Х. (октябрь 1974 г.). «Дегенерация немиелинизированных аксонов после перерезки нерва: ультраструктурное исследование». Журнал нейроцитологии. 3 (4): 497–512. Дои:10.1007 / BF01098736. PMID 4436692. S2CID 37385200.
- ^ Араки Т., Милбрандт Дж. (Август 1996 г.). «Нинджурин, новая молекула адгезии, индуцируется повреждением нерва и способствует росту аксонов». Нейрон. 17 (2): 353–61. Дои:10.1016 / S0896-6273 (00) 80166-X. PMID 8780658. S2CID 12471778.
- ^ Перри В.Х., Браун М.С., Цао Дж. У. (1 октября 1992 г.). «Эффективность гена, который замедляет скорость валлеровской дегенерации у мышей C57BL / Ola, снижается с возрастом». Европейский журнал нейробиологии. 4 (10): 1000–2. Дои:10.1111 / j.1460-9568.1992.tb00126.x. PMID 12106435.
- ^ Перри, В. Х., Ланн, Э. Р., Браун, М. К., Каузак, С. и Гордон, С. (1990), Доказательства того, что скорость валлеровской дегенерации контролируется одним аутосомно-доминантным геном. Европейский журнал нейробиологии, 2: 408-413. https://doi.org/10.1111/j.1460-9568.1990.tb00433.x
- ^ Lyon MF, Ogunkolade BW, Brown MC, Atherton DJ, Perry VH (октябрь 1993 г.). «Ген, влияющий на дегенерацию валлеровского нерва, отображается дистально на хромосоме 4 мыши». Труды Национальной академии наук Соединенных Штатов Америки. 90 (20): 9717–20. Bibcode:1993ПНАС ... 90.9717Л. Дои:10.1073 / пнас.90.20.9717. ЧВК 47641. PMID 8415768.
- ^ а б c Араки Т., Сасаки Ю., Милбрандт Дж. (Август 2004 г.). «Повышенный ядерный биосинтез NAD и активация SIRT1 предотвращают дегенерацию аксонов». Наука. 305 (5686): 1010–3. Bibcode:2004Научный ... 305.1010A. Дои:10.1126 / science.1098014. PMID 15310905.
- ^ Мак Т.Г., Райнер М., Бейровски Б., Ми В., Эмануэли М., Вагнер Д., Томсон Д., Гиллингвотер Т., Корт Ф., Конфорти Л., Фернандо Ф.С., Тарлтон А., Андрессен С., Аддикс К., Магни Дж., Рибчестер Р. Р., Перри В. Х. , Coleman MP (декабрь 2001 г.). «Валлеровская дегенерация поврежденных аксонов и синапсов задерживается химерным геном Ube4b / Nmnat». Природа Неврология. 4 (12): 1199–206. Дои:10.1038 / nn770. HDL:1842/737. PMID 11770485. S2CID 8316115.
- ^ Бейровски Б., Бабетто Е., Джилли Дж., Маццола Ф., Конфорти Л., Янецкова Л., Магни Дж., Рибчестер Р. Р., Колман депутат (январь 2009 г.). «Неядерный Wld (S) определяет его нейрозащитную эффективность для аксонов и синапсов in vivo». Журнал неврологии. 29 (3): 653–68. Дои:10.1523 / JNEUROSCI.3814-08.2009. ЧВК 6665162. PMID 19158292.
- ^ Glass JD, Brushart TM, George EB, Griffin JW (май 1993 г.). «Продолжительное выживание перерезанных нервных волокон у мышей C57BL / Ola является внутренней характеристикой аксона». Журнал нейроцитологии. 22 (5): 311–21. Дои:10.1007 / BF01195555. PMID 8315413. S2CID 45871975.
- ^ Адальберт Р., Ногради А., Сабо А., Колман депутат (октябрь 2006 г.). «Ген медленной валлеровской дегенерации in vivo защищает моторные аксоны, но не их клеточные тела после отрыва и аксотомии новорожденного». Европейский журнал нейробиологии. 24 (8): 2163–8. Дои:10.1111 / j.1460-9568.2006.05103.x. PMID 17074042.
- ^ Ван Дж, Чжай Кью, Чен И, Линь Э, Гу В., Макберни М.В., Хе З (август 2005 г.). «Местный механизм опосредует НАД-зависимую защиту от дегенерации аксонов». Журнал клеточной биологии. 170 (3): 349–55. Дои:10.1083 / jcb.200504028. ЧВК 2171458. PMID 16043516.
- ^ Conforti L, Fang G, Beirowski B, Wang MS, Sorci L, Asress S, Adalbert R, Silva A, Bridge K, Huang XP, Magni G, Glass JD, Coleman MP (январь 2007 г.). «Пересмотр NAD (+) и дегенерации аксонов: Nmnat1 не может заменить Wld (S) для задержки валлеровской дегенерации». Гибель клеток и дифференциация. 14 (1): 116–27. Дои:10.1038 / sj.cdd.4401944. PMID 16645633.
- ^ Бабетто Э., Бейровски Б., Янекова Л., Браун Р., Джилли Дж., Томсон Д., Рибчестер Р. Р., Колман М. П. (октябрь 2010 г.). «Нацеливание NMNAT1 на аксоны и синапсы трансформирует его нейропротекторную способность in vivo». Журнал неврологии. 30 (40): 13291–304. Дои:10.1523 / JNEUROSCI.1189-10.2010. ЧВК 6634738. PMID 20926655.
- ^ Фуджики М., Чжан З., Гут Л., Стюард О. (июль 1996 г.). «Генетическое влияние на клеточные реакции на повреждение спинного мозга: активация макрофагов / микроглии и астроцитов задерживается у мышей, несущих мутацию (WldS), которая вызывает замедленную дегенерацию Валлера». Журнал сравнительной неврологии. 371 (3): 469–84. Дои:10.1002 / (SICI) 1096-9861 (19960729) 371: 3 <469 :: AID-CNE9> 3.0.CO; 2-0. PMID 8842900.
- ^ Браун М.С., Перри В.Х., Хант С.П., Лаппер С.Р. (март 1994 г.). «Дальнейшие исследования регенерации моторных и сенсорных нервов у мышей с замедленной валлеровской дегенерацией». Европейский журнал нейробиологии. 6 (3): 420–8. Дои:10.1111 / j.1460-9568.1994.tb00285.x. PMID 8019679.
- ^ Остерло Дж. М., Ян Дж., Руни Т. М., Фокс А. Н., Адальберт Р., Пауэлл Е. Х., Шихан А. Е., Эйвери М. А., Хакетт Р., Логан М. А., Макдональд Дж. М., Зигенфусс Д. С., Милде С., Хоу Ю. Дж., Натан С., Динг А., Браун Р. Х. , Conforti L, Coleman M, Tessier-Lavigne M, Züchner S, Freeman MR (июль 2012 г.). «dSarm / Sarm1 необходим для активации пути гибели аксонов, вызванного повреждением». Наука. 337 (6093): 481–4. Bibcode:2012Sci ... 337..481O. Дои:10.1126 / science.1223899. ЧВК 5225956. PMID 22678360.
- ^ Гердтс Дж., Саммерс Д.В., Сасаки Ю., ДиАнтонио А., Милбрандт Дж. (Август 2013 г.). «Опосредованная Sarm1 дегенерация аксона требует взаимодействия как SAM, так и TIR». Журнал неврологии. 33 (33): 13569–80. Дои:10.1523 / JNEUROSCI.1197-13.2013. ЧВК 3742939. PMID 23946415.
- ^ Ли ХК, Чжао ЙДж (2019). «Решение топологической загадки передачи сигналов Ca 2+ с помощью циклической АДФ-рибозы и NAADP». Журнал биологической химии. 294 (52): 19831–19843. Дои:10.1074 / jbc.REV119.009635. ЧВК 6937575. PMID 31672920.
- ^ а б Гердтс Дж., Брейс Э.Дж., Сасаки Й., ДиАнтонио А., Милбрандт Дж. (Апрель 2015 г.). «Активация SARM1 локально запускает дегенерацию аксонов через разрушение NAD⁺». Наука. 348 (6233): 453–7. Bibcode:2015Научный ... 348..453G. Дои:10.1126 / science.1258366. ЧВК 4513950. PMID 25908823.
- ^ а б Эссуман К., Саммерс Д.В., Сасаки Ю., Мао Х, ДиАнтонио А., Милбрандт Дж. (Март 2017 г.). «+ Активность расщепления, которая способствует патологической дегенерации аксонов». Нейрон. 93 (6): 1334–1343.e5. Дои:10.1016 / j.neuron.2017.02.022. ЧВК 6284238. PMID 28334607.
- ^ Сасаки Ю., Накагава Т., Мао X, ДиАнтонио А., Милбрандт Дж. (Октябрь 2016 г.). «+ истощение». eLife. 5. Дои:10.7554 / eLife.19749. ЧВК 5063586. PMID 27735788.
- ^ Гилли Дж., Рибчестер Р.Р., Колман депутат (октябрь 2017 г.). "S, обеспечивает спасение на всю жизнь в мышиной модели тяжелой аксонопатии". Отчеты по ячейкам. 21 (1): 10–16. Дои:10.1016 / j.celrep.2017.09.027. ЧВК 5640801. PMID 28978465.
- ^ Ян Дж., Ву З., Ренье Н., Саймон Д. Д., Урю К., Парк Д. С., Грир П. А., Турнье С., Дэвис Р. Дж., Тесье-Лавин М. (январь 2015 г.). «Патологическая гибель аксонов из-за каскада MAPK, который вызывает локальный дефицит энергии». Клетка. 160 (1–2): 161–76. Дои:10.1016 / j.cell.2014.11.053. ЧВК 4306654. PMID 25594179.
- ^ Уокер LJ, Саммерс DW, Сасаки Y, Брейс EJ, Милбрандт J, DiAntonio A (январь 2017 г.). «Передача сигналов MAPK способствует дегенерации аксонов за счет ускорения обмена фактора поддержания аксонов NMNAT2». eLife. 6. Дои:10.7554 / eLife.22540. ЧВК 5241118. PMID 28095293.
- ^ Хеннингер Н. и др. (2016). «Ослабление травматического повреждения аксонов и улучшение функционального результата после черепно-мозговой травмы у мышей без Sarm1». Мозг. 139 (4): 1094–1105. Дои:10.1093 / мозг / aww001. ЧВК 5006226. PMID 26912636.
внешняя ссылка
| Классификация |
|---|
- Валлериан + дегенерация в Национальной медицинской библиотеке США Рубрики медицинской тематики (MeSH)