Реакция Бэмфорда – Стивенса - Википедия - Bamford–Stevens reaction
В Реакция Бэмфорда – Стивенса это химическая реакция посредством чего лечение тозилгидразоны с сильной базой дает алкены.[1][2][3] Он назван в честь британского химика. Уильям Рэндалл Бэмфорд и шотландский химик Томас Стивенс Стивенс (1900–2000). Использование апротонные растворители дает преимущественно Z-алкены, пока протонный растворитель дает смесь E- и Z-алкенов. В качестве превращения с образованием алкена реакция Бэмфорда-Стивенса имеет широкое применение в методологии синтеза и синтезе сложных молекул.

Обработка тозилгидразонов алкиллитиевыми реагентами называется Реакция Шапиро.
Механизм реакции
Первой стадией реакции Бэмфорда – Стивенса является образование диазосоединения 3.[4]

В протонных растворителях диазосоединение 3 разлагается на ион карбения 5.

В апротонных растворителях диазосоединение 3 разлагается на карбен 7.

Направленная реакция Бэмфорда-Стивенса
Реакция Бэмфорда-Стивенса не оказалась полезной для стереоселективной генерации алкенов через термическое разложение металлизированных тозилгидразонов из-за неразборчивой 1,2-перегруппировки карбенового центра, которая дает смесь продуктов. Путем замены алкильной группы триметилсилильной (ТМС) группой на N-азиридинилиминах можно повысить миграцию конкретного атома водорода. С кремний атом бета в H, a σC-Si → σ*C-H стереоэлектронный эффект ослабляет связь C-H, что приводит к ее исключительной миграции и почти исключительному образованию аллилсиланов вместо равных количеств аллилсиланов и изомерных гомоаллилсиланов, аналогично смеси продуктов, наблюдаемых в случае диалкила, или других продуктов внедрения (например, циклопропанов). Видеть бета-кремний эффект.[5][6][7]

Синтез 3-замещенных индазолов из аринов и N-тозилгидразонов
N-тозилгидразоны можно использовать в различных синтетических процедурах. Их использование с аринами было использовано для получения 3-замещенных индазолов двумя предложенными путями. Первым шагом является депротонирование гидразона диазосоединений с использованием CsF. На этом этапе конъюгатное основание может либо разложиться с образованием диазосоединения, либо претерпеть диполярный [3 + 2] циклоприсоединение с Арина дать продукту, или [3 + 2] аннулирование с арином, который также дает конечный продукт. Хотя сильные базы, такие как LiOtBu и Cs2CO3 часто используются в этой химии, CsF использовался для облегчения образования аринов in situ из о- (триметилсилил) арилтрифлатов. Также считалось, что CsF является достаточно основным, чтобы депротонировать N-тозилгидразон.[8][9]
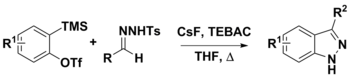
N-тозилгидразоны как реагенты для реакций кросс-сочетания
Барлуенга и его коллеги разработали первый пример использования N-тозилгидразонов в качестве нуклеофильный партнеры в реакциях кросс-сочетания. Обычно нуклеофильные реагенты в реакции сочетания как правило, из металлоорганический разновидности, а именно магнийорганический, -цинк, -олово, -кремний и -бор. В сочетании с электрофильный арилгалогениды, N-тозилгидразоны могут быть использованы для получения полизамещенных олефинов в условиях, катализируемых палладием, без использования часто дорогостоящих и сложных в синтетическом отношении металлоорганических реагентов.
Размах реакции широк; N-тозилгидразоны, полученные из альдегидов и кетонов, хорошо переносятся, что приводит к образованию как ди-, так и тризамещенных олефинов. Кроме того, различные арилгалогениды хорошо переносятся в качестве партнеров по связыванию, включая те, которые имеют как электроноакцепторные, так и электронодонорные группы, а также π-богатые и π-дефицитные ароматный гетероциклические соединения. Стереохимия - важный элемент, который следует учитывать при получении полизамещенных олефинов. Использование гидразонов, полученных из линейных альдегидов, приводило исключительно к транс-олефинам, в то время как стереохимические результаты тризамещенных олефинов зависели от размера заместителей.

Считается, что механизм этого превращения протекает аналогично синтезу алкенов посредством реакции Бэмфорда-Стивенса; разложение N-тозилгидразонов в присутствии основания с образованием диазосоединений, которые затем выделяют газообразный азот с образованием карбена, который затем можно гасить с помощью электрофила. В этом случае реакция связывания начинается с окислительная добавка арилгалогенида в Pd0 катализатор для получения арил-PdII сложный. Реакция диазосоединения, образованного из гидразона, с PdII комплекс производит комплекс Pd-карбен. А миграционная вставка арильной группы дает алкильный комплекс Pd, который претерпевает синтез устранение бета-гидрида для получения транс-арилолефина и регенерации Pd0 катализатор. Эта реакция также оказалась полезной для получения конъюгированных енинов из N-тозилгидразонов и концевых алкинов в аналогичных условиях реакции, катализируемой Pd, и по тому же механизму.

Кроме того, Барлуенга и его коллеги продемонстрировали трехкомпонентную реакцию сочетания альдегидов или кетонов, тозилгидразидов и арилгалогенидов, в которой N-тозилгидразон образуется in situ. Этот процесс дает стереоселективные олефины с выходами, аналогичными по сравнению с процессом, в котором предварительно получены N-тозилгидразоны используются.[10]
Барлуенга и его коллеги также разработали методологию безметаллового восстановительного связывания N-тозилгидразонов с бороновыми кислотами. Реакция допускает наличие множества функциональных групп на обоих субстратах, включая ароматические, гетероароматические, алифатические, электронодонорные и электроноакцепторные заместители, и протекает с высокими выходами в присутствии карбоната калия. Считается, что реакция протекает через образование диазосоединения, которое образуется из соли гидразона. Затем диазосоединение может реагировать с бороновой кислотой с образованием бензилбороновой кислоты через промежуточный боронат. Альтернативный путь состоит из образования бензилбороновой кислоты через цвиттерионный интермедиат с последующим образованием протодеборация бензилбороновой кислоты в основных условиях, что приводит к конечному восстановительному продукту.

Эта методология также была распространена на гетероатомные нуклеофилы для производства простых и тиоэфиров.[11][12]
Тандемная катализируемая родием перегруппировка Бэмфорда-Стивенса / термоалифатическая перегруппировка Клайзена
Новый процесс был разработан Штольцем, в котором реакция Бэмфорда-Стивенса была объединена с реакцией Перестановка Клейзена для производства различных олефиновых продуктов. Это превращение происходит сначала путем термического разложения N-азиридинилгидразонов с образованием диазосоединения (1) с последующим родиевым де-диазотированием (2) и син 1,2-гидридный сдвиг (3). Эта подложка подвергается термическому алифатическому Перестановка Клейзена (4), чтобы получить продукт.[13][14]

Приложение к тотальному синтезу
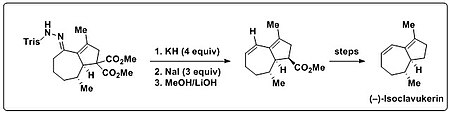
Trost et al. использовали реакцию Бэмфорда – Стивенса в своих полный синтез (-) - изоклавукерина для введения диен фрагмент, обнаруженный в натуральном продукте. Бициклический трисилгидразон изначально подвергался воздействию Реакция Шапиро условия (алкиллитий или LDA), которые приводят только к неописуемым продуктам разложения. Однако когда на этот бициклический трисилгидразон воздействовали сильным основанием (KH) и нагреванием, образовывался желаемый диеновый продукт. Более того, было показано, что образование олефинов и следующие декарбоксилирование можно было выполнить в одном горшке. С этой целью был добавлен избыток NaI, а также повышение температуры для облегчения Крапчо декарбоксилирование.[15][16]
Рекомендации
- ^ Bamford, W. R .; Стивенс, Т. С. (1952). «924. Разложение толуол-пара-сульфонилгидразонов щелочью». Журнал химического общества: 4735. Дои:10.1039 / JR9520004735.
- ^ Шапиро, Р. Х. (март 1976 г.). «Алкены из тозилгидразонов». Органические реакции. 23. Нью-Йорк: Вили. С. 405–507. ISBN 0-471-19624-X.
- ^ Adlington, R.M .; Барретт, А. Г. М. (1983). «Недавние применения реакции Шапиро». Отчеты о химических исследованиях. 16 (2): 55. Дои:10.1021 / ar00086a004.
- ^ Creary, X. (1986). «Пиролизы солей тозилгидразона: фенилдиазометаны». Органический синтез. 64: 207. Дои:10.15227 / orgsyn.064.0207. (также в Коллективный объем (1990) 7:438 (PDF))
- ^ Саркар, Т. (1992). "Кремний-направленная реакция Бэмфорда-Стивенса β-триметилсилил N-азиридинилиминов". J. Chem. Soc. Chem. Commun. (17): 1184–1185. Дои:10.1039 / C39920001184.
- ^ Ламберт, Дж. (1990). «Взаимодействие кремния с положительно заряженным углеродом». Тетраэдр. 46 (8): 2677–2689. Дои:10.1016 / s0040-4020 (01) 88362-9.
- ^ Йоргенсен, В. (1985). «Величина и происхождение эффекта β-кремния на ионы карбения». Варенье. Chem. Soc. 107 (6): 1496–1500. Дои:10.1021 / ja00292a008.
- ^ Фэн, С. (2011). «Синтез 3-замещенных индазолов из аринов и N-тозилгидразонов». Орг. Латыш. 13 (13): 3340–3343. Дои:10,1021 / ol201086g. PMID 21630698.
- ^ Пеллиссье, Х. (2002). «Использование аринов в органическом синтезе». Тетраэдр. 59 (6): 701–730. Дои:10.1016 / s0040-4020 (02) 01563-6.
- ^ Баруенга, Дж. (2007). «N-тозилгидразоны как реагенты для реакций перекрестного связывания: путь к полизамещенным олефинам». Энгью. Chem. Int. Эд. 46 (29): 5587–5590. Дои:10.1002 / anie.200701815. PMID 17577897.
- ^ Чжихуэй, С. (2012). «N-Тозилгидразоны: универсальные реагенты для реакций кросс-сочетания, катализируемых и не содержащих металлов». Chem. Soc. Rev. 41 (2): 560–572. Дои:10.1039 / c1cs15127d. PMID 21785803.
- ^ Барлуенга, Дж. (2009). «Безметалловая углерод-углеродная восстановительная связь между борными кислотами и тозилгидразонами». Nat. Chem. 1 (6): 494–499. Bibcode:2009НатЧ ... 1..494Б. Дои:10.1038 / nchem.328. PMID 21378917. S2CID 35892518.
- ^ Штольц, Б. (2002). "Некарбонилстабилизированные металлокарбеноиды в синтезе: разработка тандемно-катализируемой родием последовательности перегруппировки Бэмфорда-Стивенса / термической алифатической перегруппировки Клейзена" (PDF). Варенье. Chem. Soc. 124 (42): 12426–12427. Дои:10.1021 / ja028020j. PMID 12381180.
- ^ Вуд, Дж. (1999). «Разработка инициированной карбеноидом родия перегруппировки Клайзена для энантиоселективного синтеза α-гидроксикарбонильных соединений». Варенье. Chem. Soc. 121 (8): 1748–1749. Дои:10.1021 / ja983294l.
- ^ Трост, Б. (1996). "О диастереоселективности внутримолекулярных Pd-катализируемых циклоприсоединений ТММ. Асимметричный синтез пергидроазулена (-) - изоклавукерина А". Варенье. Chem. Soc. 118 (42): 10094–10105. Дои:10.1021 / ja961561m.
- ^ Kurti, L .; Чако, Б. (2005). Стратегические применения названных реакций в органическом синтезе. Эль-Севьер. ISBN 978-0124297852.