Олефинирование Петерсона - Peterson olefination
| Олефинирование Петерсона | |
|---|---|
| Названный в честь | Дональд Джон Петерсон |
| Тип реакции | Реакция сцепления |
| Идентификаторы | |
| Портал органической химии | Петерсон-олефинирование |
| RSC ID онтологии | RXNO: 0000080 |
В Олефинирование Петерсона (также называемый Реакция Петерсона) это химическая реакция α-силильных карбанионов (1 на диаграмме ниже) с кетоны (или же альдегиды ) с образованием β-гидроксисилана (2), что исключает образование алкены (3).[1]

Опубликовано несколько обзоров.[2][3][4][5][6]
Механизм реакции
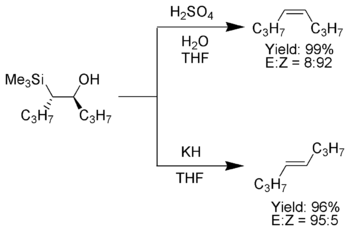
Одной из привлекательных особенностей олефинирования Петерсона является то, что его можно использовать для получения цис- или транс-алкенов из одного и того же β-гидроксисилана. Обработка β-гидроксисилана кислотой дает один алкен, а обработка того же β-гидроксисилана основанием дает алкен противоположной стереохимии.
Основное устранение
Действие основания на β-гидроксисилан (1) приводит к согласованному син устранение (2) или же (3) с образованием желаемого алкена. Пента-координата силикат средний (3) постулируется, но на сегодняшний день нет никаких доказательств.[когда? ]
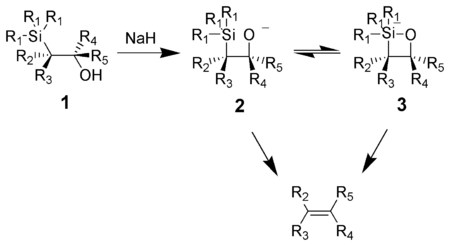
Калий алкоксиды быстро удаляются, а натрий алкоксиды обычно требуют нагревания. Магний алкоксиды удаляют только в экстремальных условиях. Порядок реакционной способности алкоксидов, K> Na >> Mg, соответствует более высокой электронной плотности на кислород, следовательно, увеличивая нуклеофильность алкоксида.
Кислотное устранение
Обработка β-гидроксисилана (1) с кислотой приводит к протонированию и анти удаление с образованием желаемого алкена.

Алкильные заместители
Когда α-силилкарбанион содержит только алкил, водород, или же электронодонорные заместители, то стереохимический результат олефинирования Петерсона можно контролировать,[7] потому что при низкой температуре удаление происходит медленно, и промежуточный β-гидроксисилан может быть выделен.
После выделения разделяют диастереомерные β-гидроксисиланы. Один диастереомер обрабатывают кислотой, в то время как другой обрабатывают основанием, таким образом превращая материал в алкен с необходимой стереохимией.[4]
Электроноакцепторные заместители
Когда α-силилкарбанион содержит электроноакцепторные заместители, олефинирование Петерсона непосредственно образует алкен. Промежуточный β-гидроксисилан нельзя выделить, так как он устраняет на месте. В этих случаях постулируется основной путь элиминации.
Вариации
Условия кислотного отщепления иногда невозможны, поскольку кислота также способствует образованию двойной связи. изомеризация. Кроме того, устранение с помощью натрий или же гидрид калия может быть невозможно из-за несовместимости функциональные группы. Чан и другие. обнаружили, что ацилирование промежуточного силилкарбинола либо ацетилхлорид или же тионилхлорид дает β-силил сложный эфир который самопроизвольно удалится при 25 ° C с образованием желаемого алкена.[8] Кори и его сотрудники разработали метод (иногда называемый Олефинирование Кори-Петерсона[9]) с использованием силилированного имина с получением α, β-ненасыщенного альдегида из карбонильного соединения в одну стадию.[10] Пример его использования в полном синтезе см .: Общий синтез таксола Kuwajima
Смотрите также
Рекомендации
- ^ Д. Дж. Петерсон (1968). «Реакция карбонилолефинирования с использованием силилзамещенных металлоорганических соединений». J. Org. Chem. 33 (2): 780–784. Дои:10.1021 / jo01266a061.
- ^ Биркофер, Л .; Штиль, О. Вершина. Curr. Chem. 1980, 88, 58. (Обзор)
- ^ Агер, Д. Дж. Синтез 1984, 384–398. (Рассмотрение)
- ^ а б Агер, Д. Дж. Орг. Реагировать. 1990, 38, 1. Дои:10.1002 / 0471264180.or038.01
- ^ Т. Х. Чан (1977). «Синтез алкенов через β-функционализированные кремнийорганические соединения». Соотв. Chem. Res. 10 (12): 442–448. Дои:10.1021 / ar50120a003.
- ^ Новые разработки в реакции олефинирования Петерсона L. Frances van Staden, David Gravestock и David J. Ager Chem. Soc. Ред., 2002, 31, 195-200 Дои:10.1039 / A908402I
- ^ Barrett, A.G.M .; Flygare, J. A .; Hill, J.M .; Уоллес, Э. М. (1998). «Стереоселективный синтез алкенов через 1-хлор-1 - [(диметил) фенилсилил] алканы и α- (диметил) фенилсилилкетоны: 6-метил-6-додецен». Органический синтез.; Коллективный объем, 9, п. 580
- ^ Т. Х. Чан и Э. Чанг (1974). «Синтез алкенов из карбонильных соединений и альфа карбанионов в кремний. III. Полный отчет и синтез полового феромона непарной моли». J. Org. Chem. 39 (22): 3264–3268. Дои:10.1021 / jo00936a020. PMID 4473100.
- ^ X. Zeng; Ф. Цзэн и Э. Негиши (2004). «Эффективный и селективный синтез 6,7-дегидростипиамида посредством Zr-катализируемого асимметричного карбоалюминирования и Pd-катализируемого перекрестного связывания оцинкованных органических соединений». Орг. Lett. 6 (19): 3245–3248. Дои:10.1021 / ol048905v. PMID 15355023.
- ^ Э. Дж. Кори; Д. Эндерс и М. Г. Бок (1976). «Простой и высокоэффективный путь к α-β-ненасыщенным альдегидам». Буквы Тетраэдра. 17 (1): 7–10. Дои:10.1016 / S0040-4039 (00) 71308-6.