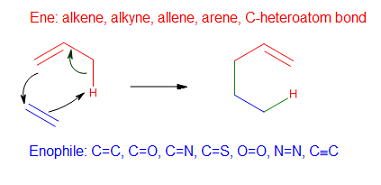
Эне реакция - Ene reaction
В ее реакция (также известный как Ольдереновая реакция его первооткрыватель Курт Алдер в 1943 г.) является химическая реакция между алкен с аллильный водород (в ene) и соединение, содержащее кратную связь ( энофил), чтобы образовать новую σ-связь с миграцией еновой двойной связи и 1,5-водородным сдвигом. Продукт представляет собой замещенный алкен с двойной связью, смещенной в аллильное положение.[1]
Это преобразование - групповой перенос перициклическая реакция,[2] и поэтому обычно требуются высокоактивированные подложки и / или высокие температуры.[3] Тем не менее, реакция совместима с широким спектром функциональные группы которые могут быть присоединены к еновому и енофильному фрагментам. Много полезного Кислота Льюиса Были также разработаны катализированные еновые реакции, которые могут обеспечить высокие выходы и селективность при значительно более низких температурах, что делает еновую реакцию полезным инструментом образования C – C для синтеза сложных молекул и природных продуктов.
Компонент Ene
Ены представляют собой π-связанные молекулы, которые содержат по крайней мере один активный атом водорода в аллильном, пропаргильном или α-положении. Возможные еновые компоненты включают олефиновые, ацетиленовые, алленовые, ароматические, циклопропильные и углерод-гетеро-связи.[4] Обычно аллильный водород алленовых компонентов участвует в еновых реакциях, но в случае алленилсиланов алленовый атом водорода α на кремниевый заместитель переносится, давая силилалкин. Фенол может действовать как еновый компонент, например, в реакции с дигидропираном, но требуются высокие температуры (150–170 ° C). Тем не менее напряженные ены и конденсированные малые кольцевые системы вступают в еновые реакции при гораздо более низких температурах. Кроме того, сообщалось о компонентах ена, содержащих связи C = O, C = N и C = S, но такие случаи редки.[4]
Энофил
Энофилы представляют собой молекулы с π-связями, которые имеют электроноакцепторные заместители, которые значительно снижают LUMO π-связи. Возможные энофилы содержат кратные связи углерод-углерод (олефины, ацетилены, бензины), кратные углерод-гетеро-связи (C = O в случае карбониленовых реакций, C = N, C = S, C≡P), гетеро-гетеро кратные связи (N = N, O = O, Si = Si, N = O, S = O), кумулен системы (N = S = O, N = S = N, C = C = O, C = C = S, SO2) и заряженных π-систем (C = N+, C = S+, C≡O+, C≡N+).[4]
Ретро-еновая реакция
Обратный процесс, обратная реакция, может иметь место при экструзии термодинамически стабильных молекул, таких как диоксид углерода или диазот. Например, кинетические данные и компьютерные исследования показывают, что термолиз бут-3-еновой кислоты с образованием пропена и диоксида углерода происходит по ретроеновому механизму.[5] Точно так же пропаргиловые диазены легко разлагаются по ретроеновому механизму с образованием алленовых продуктов и газообразного азота (см. Синтез аллен Майерс ).
Механизм
Согласованные пути и переходные состояния
Основное погранично-орбитальное взаимодействие, происходящее в еновой реакции, происходит между HOMO ена и НСМО энофила (рис. 2).[6] ВЗМО ена является результатом комбинации пи-связывающей орбитали в виниловом фрагменте и связывающей СН орбитали для аллильного Н. В целом, полностью карбон-еновые реакции имеют, как правило, высокий активационный барьер, который был приблизительно равен при 138 кДж / моль в случае пропена и этена, как вычислено на уровне теории M06-2X / def2-TZVPP.[7] Однако, если энофил становится более полярным (переходя от этана к формальдегиду), его LUMO имеет большую амплитуду на C, что дает лучшее перекрытие C – C и худшее перекрытие H – O, определяя, что реакция протекает асинхронно. Это приводит к снижению активационного барьера до 61,5 кДж / моль (M06-2X / def2-TZVPP), если S заменяет O на энофиле. Путем компьютерного исследования как барьеров активации, так и штаммов активации нескольких различных еновых реакций с участием пропена в качестве енового компонента, Фернандес с соавторами [7] обнаружили, что барьер уменьшается вдоль энофилов в порядке H2C = CH2 > H2C = NH> H2C = CH (COOCH3)> H2С = О> Н2C = PH> H2C = S, поскольку реакция становится все более и более асинхронной и / или напряжение активации уменьшается.

Согласованность процесса ene подтверждена экспериментально,[8] и реакцию можно обозначить как [σ2s + π2s + π2s] в обозначениях Вудворда-Гофмана.[6] Раннее переходное состояние, предложенное для термической реакции пропена с формальдегидом, имеет конформацию оболочки с углом C – O – H 155 °, как рассчитано на уровне теории 3–21G.[9]
Шнабель и его сотрудники[10] исследовали некаталитическую внутримолекулярную карбониленовую реакцию, которая была использована для получения циклопентанового фрагмента природных и неприродных ятрофа-5,12-диенов, членов семейства модуляторов P-гликопротеина. Их расчеты методом DFT на уровне теории B1B95 / 6-31G * для реакции, представленной на рисунке 3, предполагают, что реакция может протекать через одно из двух конкурирующих согласованных переходных состояний, подобных оболочке. Развитие 1,3-трансаннулярных взаимодействий в неблагоприятном переходном состоянии дает хорошее объяснение избирательности этого процесса.
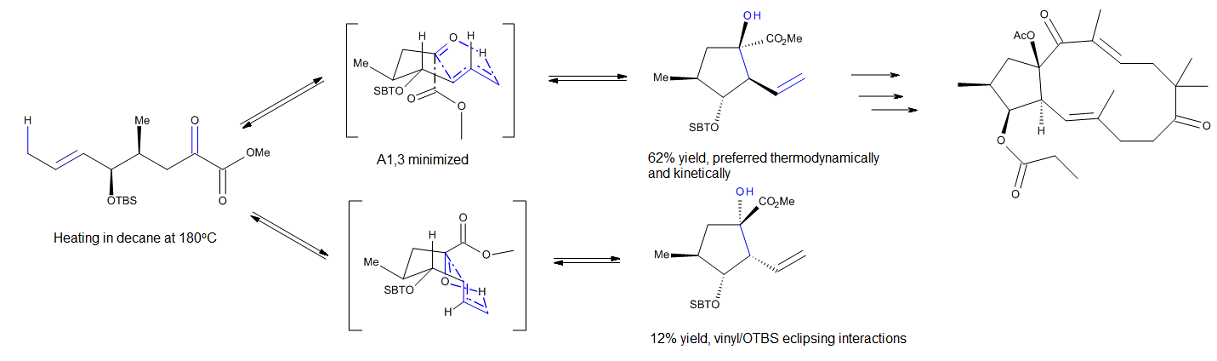
Изучение кислоты Льюиса, способствующей карбониленовым реакциям, таким как глиоксилат-еновые процессы, катализируемые алюминием (рис. 4), побудило исследователей рассмотреть конформацию, подобную стулу, для переходного состояния еновых реакций, которые протекают с относительно поздними переходными состояниями.[2] Преимущество такой модели заключается в том, что стерические параметры, такие как 1,3-диаксиальное и 1,2-диэкваториальное отталкивание, легко визуализировать, что позволяет делать точные прогнозы относительно диастереоселективности многих реакций.[2]

Радикальный механизм
Когда согласованный механизм геометрически невыгоден, термическая реакция может протекать по ступенчатому бирадикальному пути.[11] Другая возможность - свободнорадикальный процесс, если радикальный инициаторы присутствуют в реакционной смеси. Например, еновая реакция циклопентена и циклогексена с диэтилазодикарбоксилат может катализироваться радикальными инициаторами. Как видно на рисунке 5, ступенчатому характеру процесса способствует стабильность циклопентенильного или циклогексенильного радикалов, а также сложность циклопентен и циклогексен в достижении оптимальной геометрии для согласованного процесса.[12][требуется разъяснение ]

Региоселекция
Как и в случае любого циклоприсоединения, успех еновой реакции во многом определяется стерической доступностью еналлильного водорода. В общем, атомы водорода метила и метилена отщепляются намного легче, чем атомы водорода метина. В тепловых еновых реакциях порядок реакционной способности оторванного атома H является первичным> вторичным> третичным, независимо от термодинамической стабильности внутреннего олефинового продукта. В реакциях, промотируемых кислотой Льюиса, используемая пара энофил / кислота Льюиса в значительной степени определяет относительную легкость отщепления метильных атомов водорода от метиленовых.[2]
Ориентацию присоединения ена можно предсказать по относительной стабилизации развивающихся частичных зарядов в несимметричном переходном состоянии с ранним образованием σ-связи. Главный региоизомер будет происходить из переходного состояния, в котором кратковременные заряды лучше всего стабилизируются ориентацией ена и енофила.[4]
Внутренняя асимметричная индукция
Что касается диастереоселекции по отношению к вновь созданным хиральным центрам, предпочтение эндо было качественно обнаружено, но стерические эффекты могут легко изменить это предпочтение (Рисунок 6).[2]

Внутримолекулярные еновые реакции
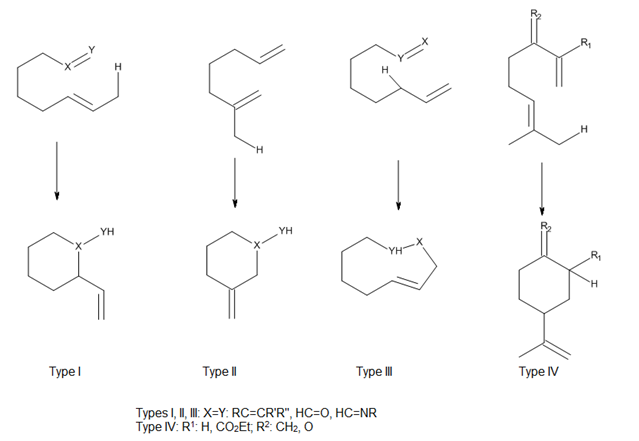
Внутримолекулярные еновые реакции выигрывают от менее отрицательной энтропии активации, чем их межмолекулярные аналоги, поэтому обычно они более легкие, протекая даже в случае простых энофилов, таких как неактивированные алкены и алкины.[13] Высокие регио- и стереоселективности, которые могут быть получены в этих реакциях, могут обеспечить значительный контроль при синтезе сложных кольцевых систем.
Учитывая положение крепления троса, соединяющего ен и энофил, Оппольцер[2] классифицировал как термические, так и катализируемые кислотой Льюиса внутримолекулярные еновые реакции как типы I, II и III, а Snider[3] добавил реакцию типа IV (рис. 7). В этих реакциях перекрытие орбиталей ена и энофила в значительной степени контролируется геометрией сближения компонентов.[4]
Кислота Льюиса - катализируемые еновые реакции
Преимущества и обоснование
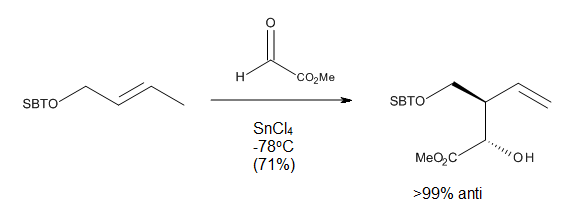
Термические реакции имеют несколько недостатков, таких как необходимость очень высоких температур и возможность побочных реакций, таких как протонно-катализируемая полимеризация олефинов или реакции изомеризации. Поскольку энофилы электронодефицитны, было высказано предположение, что их комплексообразование с кислотами Льюиса должно ускорять реакцию ена, как это происходило для реакции, показанной на рисунке 8.

Галогениды алкилалюминия хорошо известны как поглотители протонов, и их использование в качестве катализаторов на основе кислот Льюиса в еновых реакциях значительно расширило объем этих реакций и позволило их изучить и разработать в значительно более мягких условиях.[3]
Поскольку кислота Льюиса может непосредственно образовывать комплекс с карбонильным кислородом, для энофилов, содержащих связь C = O, были разработаны многочисленные катализаторы на основе триалкилалюминия. В частности, было установлено, что Me2AlCl является очень полезным катализатором еновых реакций α, β-ненасыщенных альдегидов и кетонов, а также других алифатических и ароматических альдегидов. Причина успеха этого катализатора заключается в том, что ен-аддукт-Me2Комплекс AlCl может далее реагировать с образованием метана и алкоксида алюминия, что может предотвратить катализируемые протонами перегруппировки и сольволиз (рис. 9).[3]

В случае направленных карбониленовых реакций при добавлении кислоты Льюиса наблюдались высокие уровни регио- и стереоселективности, что можно объяснить переходными состояниями типа кресла. Некоторые из этих реакций (рис. 10) могут протекать при очень низких температурах и по-прежнему давать очень хорошие выходы одного региоизомера.[2]
Условия реакции
Пока нуклеофильность алкильной группы не приводит к побочным реакциям, каталитических количеств кислоты Льюиса достаточно для многих еновых реакций с реакционноспособными энофилами. Тем не менее количество кислоты Льюиса может широко варьироваться, так как оно в значительной степени зависит от относительной основности энофила и аддукта ена. Что касается выбора растворителя для реакций, самые высокие скорости обычно достигаются при использовании галогенуглеродов в качестве растворителей; полярные растворители, такие как простые эфиры, не подходят, поскольку они могут образовывать комплекс с кислотой Льюиса, делая катализатор неактивным.[3]
Реакционная способность енов
В то время как стерические эффекты все еще важны для определения результата реакции ена, катализируемой кислотой Льюиса, электронные эффекты также значительны, поскольку в такой реакции на центральном атоме углерода ена образуется значительный положительный заряд. В результате алкены, содержащие по крайней мере один дизамещенный виниловый углерод, намного более реакционноспособны, чем моно- или 1,2-дизамещенные.[3]
Механизм
Как видно на рисунке 11, еновые реакции, катализируемые кислотой Льюиса, могут протекать либо по согласованному механизму, который имеет полярное переходное состояние, либо по ступенчатому механизму с цвиттерионным промежуточным соединением. Ен, энофил и выбор катализатора могут влиять на то, какой путь является процессом с более низкой энергией. В общем, чем более реакционноспособен комплекс ен или энофил-кислота Льюиса, тем более вероятно, что реакция будет ступенчатой.[3]

Хиральные кислоты Льюиса для асимметричного катализа карбониленовых реакций
Хиральные диалкоксититановые комплексы и синтез лаулималида
Текущее направление в изучении еновых реакций, катализируемых кислотой Льюиса, - это разработка асимметричных катализаторов образования связи C – C. Миками [14] сообщил об использовании хирального комплекса титана (рис. 12) в асимметричных еновых реакциях с участием прохиральных эфиров глиоксилата. Катализатор получают in situ из (i-PrO)2TiX2 и оптически чистый бинафтол, причем обмен алкоксилигандом облегчается за счет использования молекулярных сит. Этот метод позволяет получать α-гидроксиэфиры высокой энантиомерной чистоты, соединения, которые представляют класс биологической и синтетической значимости (рис. 12).[14]
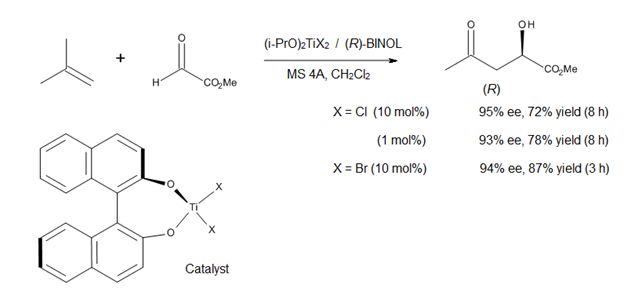
Поскольку и (R) -, и (S) -БИНОЛ коммерчески доступны в оптически чистой форме, этот асимметричный процесс позволяет синтезировать как энантиомеры α-гидроксиэфиров, так и их производные. Однако этот метод применим только к 1,1-дизамещенным олефинам из-за умеренной льюисовской кислотности комплекса титан-BINOL.[14]
Как показано на рисунке 13, Кори и его сотрудники[15] предложили раннее переходное состояние для этой реакции с целью объяснения наблюдаемой высокой энантиоселективности (предполагая, что реакция является экзотермической, как рассчитано из стандартных энергий связи). Даже если структура активного катализатора неизвестна, модель Кори предлагает следующее: альдегид активируется комплексообразованием с хиральным катализатором (R) -BINOL-TiX.2 неподеленной парой электронов формила, синфазной с водородом формила, с образованием пентакоординированной структуры Ti. Водородная связь CH-O происходит со стереоэлектронно наиболее подходящей неподеленной парой кислорода лиганда BINOL. В такой структуре верхняя (обратная) поверхность формильной группы намного более доступна для нуклеофильной атаки, поскольку нижняя (си) поверхность защищена соседним нафтольным фрагментом, что обеспечивает наблюдаемую конфигурацию продукта.

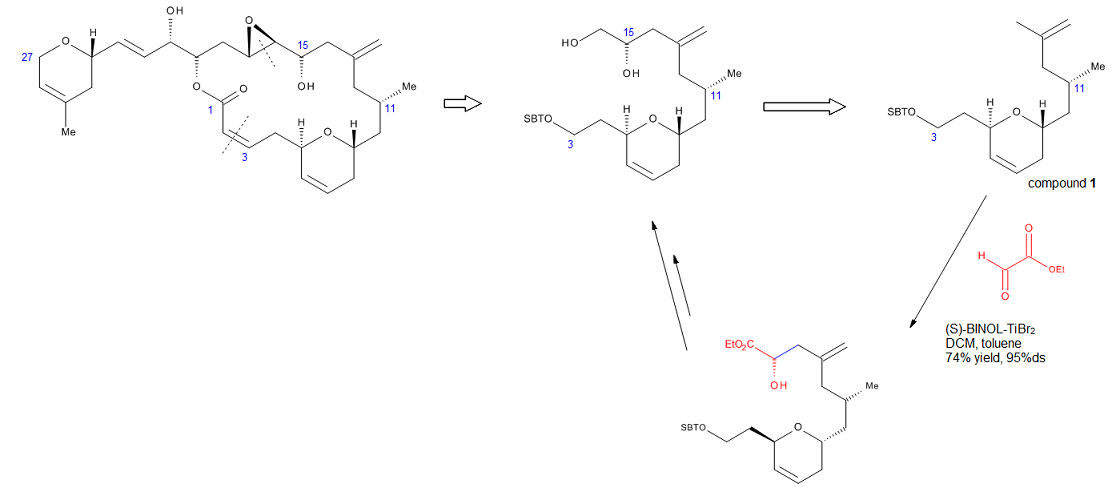
Формальный полный синтез лаулималида[16] (Рисунок 14) иллюстрирует устойчивость реакции, разработанной Миками. Лаулималид - это морской натуральный продукт, метаболит различных губок, который может найти потенциальное применение в качестве противоопухолевого средства из-за его способности стабилизировать микротрубочки. Одним из ключевых шагов в стратегии, используемой для синтеза фрагмента C3-C16, была хирально катализируемая еновая реакция, которая установила стереоцентр C15. Обработка концевой аллильной группы соединения 1 с помощью этилглиоксилат в присутствии каталитического (S) -БИНОЛ-TiBr2 обеспечивал требуемый спирт с выходом 74% и ds> 95%. Этот метод устраняет необходимость в защитной группе или любой другой функциональной группе на конце молекулы. Кроме того, проводя эту реакцию, Pitts et al. удалось избежать жестких условий и низких выходов, связанных с установкой экзо-метиленовых единиц на поздних стадиях синтеза.[16]
Хиральные C2-симметричные комплексы Cu (II) и синтез (+) - азаспирацида-1
Эванс и сотрудники [17] разработали новый тип энантиоселективных C2-симметричных Cu (II) катализаторов, в которых субстраты могут образовывать хелат через две карбонильные группы. Было обнаружено, что катализаторы обеспечивают высокие уровни асимметричной индукции в нескольких процессах, включая еновую реакцию этилглиоксилата с различными неактивированными олефинами. На рисунке 15 показаны три катализатора, которые, как они обнаружили, являются наиболее эффективными для получения гамма-дельта-ненасыщенных альфа-гидроксиэфиров с высокими выходами и превосходной энантиоселективностью. Особенностью соединения 2 является то, что он устойчив в лабораторных условиях и может храниться неограниченное время, что делает его удобным в использовании. Реакция имеет широкий диапазон, как показано на фиг. 16, благодаря высокой кислотности катализаторов по Льюису, которые могут активировать даже слабонуклеофильные олефины, такие как 1-гексен и циклогексен.


В случае катализаторов 1 и 2 было высказано предположение, что асимметричная индукция катализаторами является результатом образования плоско-квадратного комплекса катализатор-глиоксилат (Фиг.17), в котором поверхность Re альдегида блокируется трет. -бутильные заместители, позволяющие поступающим олефинам атаковать только Si-поверхность.[18] Однако эта модель не учитывает индукцию, наблюдаемую при использовании катализатора 3. Текущий вид[19] заключается в том, что геометрия металлического центра становится тетраэдрической, так что стерически экранированная грань альдегидного фрагмента является гранью Re.

Первоначально ценность метода, разработанного Эвансом и соавторами, была доказана успешным преобразованием полученного альфа-гидроксиэфира в соответствующий метиловый эфир, свободную кислоту, Вайнреб амид и сложный альфа-азидоэфир без какой-либо рацемизации, как показано на Фигуре 18.[17] Азидное замещение спирта, возникающее в результате карбониленовой реакции, обеспечивает легкий путь к синтезу ортогонально защищенных аминокислот.

Синтетическая полезность хиральных C2-симметричных Cu (II) катализаторов была действительно выявлена в образовании C17 стереоцентра кольцевого фрагмента CD (+) - азаспирацида-1, очень мощного токсина (цитотоксичного для клеток млекопитающих), продуцируемого в в незначительных количествах различных видов моллюсков, включая мидии, устрицы, гребешки, моллюски и моллюски.[20] Как показано на рисунке 19, реакция, которая устанавливает стереоцентр C17, катализируется 1 мол.% Комплекса Cu (II) 2 (рисунок 15), и авторы отмечают, что ее можно проводить в масштабе 20 г и все же давать очень хорошие выходы. и отличная энантиоселективность. Кроме того, продукт можно легко превратить в соответствующий амид Вайнреба без потери селективности, что позволяет легко вводить метильную группу C14. Таким образом, этот новый каталитический энантиоселективный процесс, разработанный Эвансом и его сотрудниками, может быть легко интегрирован в сложные проекты синтеза, особенно на ранних этапах синтеза, когда высокие выходы и энантиоселективиты имеют первостепенное значение.

Смотрите также
- Реакция Дильса-Альдера
- Определенный изотолуолы изомеризоваться по еновому механизму
Рекомендации
- ^ Ольха, К .; Пашер, Ф; Schmitz, A. "Убер умереть Anlagerung von Maleinsäure-anhydrid und Azodicarbonsäure-ester an einfach ungesättigte Koh an einfach ungesättigte Kohlenwasserstoffe. Zur Kenntnis von Substitutionsvorgängen in der Allyl-Stellung". Бер. Dtsch. Chem. Ges. 7: 2. Дои:10.1002 / cber.1943076010.
- ^ а б c d е ж грамм Миками, К .; Симидзу, М. (1992). «Асимметричные еновые реакции в органическом синтезе». Chem. Rev. 92 (5): 1021. Дои:10.1021 / cr00013a014.
- ^ а б c d е ж грамм Снайдер, Б. Б. (1980). «Катализируемые кислотой Льюиса еновые реакции». Соотв. Chem. Res. 13 (11): 426. Дои:10.1021 / ar50155a007.
- ^ а б c d е Падерес, Г. Д .; Йоргенсен, В. Л. (1992). «Компьютерная механистическая оценка органических реакций. 20. Эне и ретро-химия». J. Org. Chem. 57 (6): 1904. Дои:10.1021 / jo00032a054. и ссылки в нем
- ^ Дьюар, Майкл Дж. С .; Форд, Джордж П. (1977-12-01). «Термическое декарбоксилирование бут-3-еновой кислоты. Расчеты параметров активации и первичных кинетических изотопных эффектов MINDO / 3». Журнал Американского химического общества. 99 (25): 8343–8344. Дои:10.1021 / ja00467a049. ISSN 0002-7863.
- ^ а б Inagaki, S .; Fujimoto, H; Фукуи, К. Дж. (1976). «Орбитальное взаимодействие в трех системах». Варенье. Chem. Soc. 41 (16): 4693. Дои:10.1021 / ja00432a001.
- ^ а б Fernandez, I .; Бикельгаупт, Ф. М. (2012). «Альдереновая реакция: ароматичность и активационно-штаммовый анализ». Журнал вычислительной химии. 33 (5): 509–516. Дои:10.1002 / jcc.22877. PMID 22144106.
- ^ Stephenson, L.M .; Маттерн, Д. Л. (1976). «Стереохимия еновой реакции диметилазодикарбоксилата». J. Org. Chem. 41 (22): 3614. Дои:10.1021 / jo00884a030.
- ^ Loncharich, R.J .; Хоук, К. Н. (1987). «Переходные структуры еновых реакций этилена и формальдегида с пропеном». Варенье. Chem. Soc. 109 (23): 6947. Дои:10.1021 / ja00257a008.
- ^ Шнабель, Кристоф; Стерц, Катя; Мюллер, Хенрик; Ребейн, Юлия; Визе, Майкл; Хирсеманн, Мартин (2011). «Полный синтез природных и неприродных Δ5,6Δ12,13-ятрофан дитерпенов и их оценка как модуляторы MDR». Журнал органической химии. 76 (2): 512. Дои:10.1021 / jo1019738. PMID 21192665.
- ^ Хоффманн, Х. М. Р. (1969). "Эне реакция". Энгью. Chem. Int. Эд. 8 (8): 556. Дои:10.1002 / anie.196905561.
- ^ Thaler, W. A .; Франц, Б. Дж. (1964). «Реакция этилазодикарбоксилата с моноолефинами». J. Org. Chem. 29 (8): 2226. Дои:10.1021 / jo01031a029.
- ^ Oppolzer, W .; Сниецкус В. (1978). «Внутримолекулярные Ene реакции в органическом синтезе». Энгью. Chem. Int. Эд. Англ. 17 (7): 476. Дои:10.1002 / anie.197804761.
- ^ а б c Миками, К .; Terada, M .; Такеши, Н. (1990). «Каталитическая асимметричная глиоксилат-еновая реакция: практический доступ к & alpha; -гидроксиэфирам с высокой энантиомерной чистотой». Варенье. Chem. Soc. 112 (10): 3949. Дои:10.1021 / ja00166a035.
- ^ Кори, E.J .; Barnes-Seeman, D .; Lee, T. W .; Гудман, С. Н. (1997). «Модель переходного состояния для энантиоселективной реакции миками ен». Буквы Тетраэдра. 37 (37): 6513. Дои:10.1016 / S0040-4039 (97) 01517-7.
- ^ а б Pitts, M. R .; Мульцер, Дж. (2002). «Хирально катализируемая еновая реакция в новом формальном полном синтезе противоопухолевого агента лаулималида». Буквы Тетраэдра. 43 (47): 8471. Дои:10.1016 / S0040-4039 (02) 02086-5.
- ^ а б Evans, D.A .; Tregay, S.W .; Burgey C. S .; Paras, N.A .; Войковский, Т. (2000). «C2-симметричные комплексы меди (II) в виде хиральных кислот Льюиса. Каталитические энантиоселективные реакции карбонил-эна с глиоксилатными и пируватными эфирами». Варенье. Chem. Soc. 122 (33): 7936. Дои:10.1021 / ja000913t.
- ^ Johnson, J. S .; Эванс, Д. А. (2000). «Хиральные бис (оксазолин) комплексы меди (II): универсальные катализаторы для энантиоселективного циклоприсоединения, реакции Альдола, Михаэля и карбонилена». Соотв. Chem. Res. 33 (6): 325–35. Дои:10.1021 / ar960062n. PMID 10891050.
- ^ Йоханнсен, Могенс; Йоргенсен, Карл Анкер (1995). «Асимметричные гетеро реакции Дильса-Альдера и еновые реакции, катализируемые хиральными комплексами меди (II)». Журнал органической химии. 60 (18): 5757. Дои:10.1021 / jo00123a007.
- ^ Эванс, Д. А .; Kaerno, L .; Dunn, T. B .; Beauchemin, A .; Raymer, B .; Mulder, J. A .; Olhava, E. J .; Juhl, M .; Кагечика, К .; Фавор Д. А. (2008). «Полный синтез (+) - азаспирацида-1. Выставка тонкостей синтеза сложных молекул». Варенье. Chem. Soc. 130 (48): 16295–16309. Дои:10.1021 / ja804659n. ЧВК 3408805. PMID 19006391.