Общий синтез галантамина - Galantamine total synthesis

Статья касается полный синтез из галантамин, а препарат, средство, медикамент используется для лечения от легкой до умеренной Болезнь Альцгеймера.[1]
Естественным источником галантамина являются некоторые виды нарцисс и потому, что этих видов мало и потому что выделение галантамина из нарциссов обходится дорого (по данным 1996 г. Доллар США на килограмм урожайность нарциссов составляет 0,1–0,2% от сухого веса) альтернативные синтетические источники находятся в стадии разработки с помощью полный синтез.
Контур
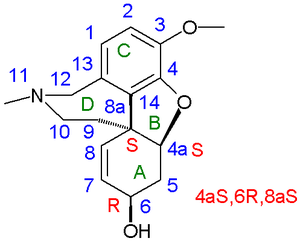
В 1962 г. рацемический галантамин и эпигалантамин были получены путем органического восстановления рацемических нарведин к Д. Х. Р. Бартон. Нарведин родственный Enone (галантамин аллиловый спирт ), полученный окислительным сочетанием. Химический выход: 1,4%. Кроме того, они изолировали (-) - навардин по хиральное разрешение из смеси рацемического нарведина и 0,5 эквивалента (+) - галантамина. Таким образом, они смогли снова получить (-) галантамин восстановлением В 1976 году Каметани получил оба энантиомера галантамина, используя производное Винная кислота как хиральный растворяющий агент. В 1977 году Кога получил оба энантиомера с помощью синтез хирального пула начиная с L-тирозин [2][3] а в 1988 году Carrol оптимизировал процесс окислительного сочетания до выхода 11% на основе изованилин.
В 1989 году Влахов использовал асимметричную редукцию на биокатализ в синтезе нескольких предшественников галантамина. а в 1994 году Shieh & Carlson [4] полученный (-) - галантамин путем спонтанное разрешение его предшественника нарведина. Рацемический нарведин обрабатывали 0,01 эквивалентом (+) - галантамина, что дало выход 76%. Нарведин представляет собой рацемический конгломерат, позволяющий изолировать S, S-энантиомер от R, R-энантиомера путем простой кристаллизации. Уникальность процесса заключается в том, что оба энантиомера находятся в динамическом химическое равновесие друг с другом через общие фенол в Реакция Майкла как реакция вызванный триэтиламин.
 |
| Решение Нарведина |
|---|
В 1999 г. Джордис выполнил (-) - синтез галантамина в многокилограммовой шкале на основе химии Кэрролла и хирального разрешения Ши / Карлсона. Это станет основой для текущего промышленного производства Sanochemia (AT). В 2000 году Фелс предложил внутримолекулярный Чертовски реакция для построения основы галантамина, и в том же году Trost & Toste получили (-) - галантамин в асимметричный синтез с участием асимметричное аллильное алкилирование и внутримолекулярная реакция Хека. Улучшенные методы были опубликованы в 2002 и 2005 гг. (См. Ниже). В 2004 г. Узел получил (-) - галантамин через удаленный асимметричная индукция метод с исходным хиральным соединением D-фенилаланин.[5] Браун приготовил (-) - галантамин в 2007 г., начиная с изованилин.[6] Изованилин также использовал Магнус (2009). [7] D-глюкоза использовалась Chida (2010).[8]
О синтезе рацемического галантамина сообщил Ван в 2006 г. [9] и Сайто в 2008 году.[10]
Промышленное производство Sanochemia
Метод, описанный Джордисом в 1999 году, составляет основу промышленного производства галантамина.[11]
 |  | |
| Синтез нарведина, часть А | Синтез нарведина, часть B |
Этот метод основан на электрофильное галогенирование 3,4-диметоксибензальдегида 1 (доступно из изованилин ) с бром / уксусная кислота к броморганический 2 с последующим региоселективный деметоксилирование с серная кислота к фенол 3. Это соединение реагирует в восстановительное аминирование (борогидрид натрия ) с тирамин 4 к амин 5 который формилированный с этилформиат и муравьиная кислота в диоксан на следующем этапе для соединения 6. An окислительное сочетание фенола происходит рядом с Феррицианид калия и карбонат калия в толуол к 7. Связь C8a-C14 образуется на первом этапе, за которым следует Майкл дополнение другой фенольной группы к новообразованной Enone группа. Этап реакции создает два стереоцентры приводит к двум диастереомерный пара энантиомеры. По характеру скелета ABD желаемая пара S, S / R, R является основным продуктом, а другая пара S, R / R, S удаляется при обработке. В кетон группа защищена как кеталь 8 с 1,2-пропиленгликоль позволяя органическое восстановление к литийалюминийгидрид как бромной группы, так и формильной группы. На втором этапе кетальная группа удаляется (соляная кислота ) с образованием рацемического (S, S / R, R) нарведина 9.
Enantiopure (-) - нарведин получают с помощью метода динамического хирального разделения, впервые предложенного Ши / Карлсоном, и на заключительном этапе кетон восстанавливается до спирта с L-селекция.
 |
| восстановление (-) - нарведина до (-) - галантамина в виде бромида |
|---|
Этот последний шаг энантиоселективный получение желаемого соединения S, S, R, потому что приближение H− ограничивается Si лицо поскольку поверхность Re защищена кольцевой системой DB. Формирование S, S, S эпимер также можно избежать, поддерживая температуру реакции ниже -15 ° C.
Синтез Трост-Галантамина
В полный синтез галантамина (Трост 2005) [12] описывается следующим образом: последовательность начинается с бромирование к электрофильное ароматическое замещение из изованилин 1 к бромфенолу 2, затем путем синтеза второго интермедиата 5 реагируя диальдегид 3 в паре альдольная реакция и Реакция Хорнера – Уодсворта – Эммонса с триметилфосфоноацетатом 4. В гидроксил группа активируется как трихлорэтилкарбонат уходящая группа к 6. Следующий энантиоселективный Реакция Trost AAA происходит между бромфенолом 2 и карбонат 6 к аллиловому эфиру 7. Далее альдегид группа защищена как ацеталь в 8 и этот шаг позволяет органическое восстановление из сложный эфир группа в алкоголь 9 с ДИБА и последующая омологация этого спирта нитрил к Мицунобу -типа с использованием циангидрина ацетона в качестве источника цианида, чтобы получить 10 с последующим снятием защиты с альдегида до 11. Внутримолекулярная реакция Хека на 12 образует дигидрофурановое кольцо. Аллильное окисление к диоксид селена содержит аллиловый спирт 13 с правильной стереохимией. Альдегид реагирует с метиламин к я добываю 14 и снижение имина и нитрила ДИБАЛ-Н приводящий к закрытию кольца аминальный 15 (не изолирован) с последующим кислотным гашением дает полуаминал 16. На последнем этапе полуаминал восстанавливается до галантамина. 17 вместе с 6% эпи-изомер 18.[13]
 |
| Trost 2005 Полный синтез галантамина |
|---|
Эли Лилли / Университет Саутгемптона Синтез галантамина
Полный синтез, сообщенный Эли Лилли и Саутгемптонский университет в 2007 году также начинается с изованилина.[6] В альдегид группа в производной 1 преобразуется в его амин к восстановительное аминирование с метиламин что тогда защищенный как Группа BOC в 2. Остальная часть углеродного каркаса добавлена хиральным пропаргиловый спирт 3 (вводя стереоцентр 4a и полученный хиральный синтез из кетон с R-альпийский боран ) в Мицунобу реакция к ариловый эфир 4. В триметилсилил защитная группа удалена карбонат калия в метанол и последующие Enyne Metathesis реакция с Катализатор Граббса дает диен 5. А реакция гидроборирования – окисления обращает 5 к алкоголь 6 а внутримолекулярная реакция Хека дает трицикл 7 с алкен изомеризация и создание 8а стереоцентр с правильным стереохимия на основе хиральная индукция. В аллиловый спирт группа в 8 вводится окисление селеноксида с избытком желаемого диастереомер. Последний шаг к галантамину 9 гидроксильная группа активируется как тройной и аминогруппа как мезилат для внутримолекулярного азепин закрытие кольца через нуклеофильное замещение (с 6% эпимер формирование).
 |  | |
| Синтез галантамина 2007 A | Синтез галантамина 2007 часть B |
Ссылки и примечания
- ^ Синтез и фармакология галантамина Хосе Марко-Контеллес, Мария ду Карму Каррейрас, Каролина Родригес, Мерседес Вильярройя и Антонио Г. Гарсиа Chem. Ред.; 2006; 106 (1) pp 116–133; (Рассмотрение) Дои:10.1021 / cr040415t
- ^ Кога, Кенджи; Томиока, Киёси; Симидзу, Кимихиро; Ямада, Шун-Ичи (1977). «Подходы к биогенетическому асимметричному синтезу некоторых алкалоидов Amaryllidaceae». Гетероциклы. 6 (9): 1752. Дои:10.3987 / R-1977-09-1752.
- ^ Кога, Кенджи; Симидзу, Кимихиро; Томиока, Киёси; Ямада, Шун-Ичи (1977). "Биогенетический асимметричный синтез оптически активных алкалоидов Amaryllidaceae: (+) - и (-) - галантамин из L-тирозина". Гетероциклы. 8: 277. Дои:10.3987 / S (S) -1977-01-0277.
- ^ Асимметричное преобразование любого энантиомера нарведина посредством процесса полного самопроизвольного разделения, краткое решение синтеза (-) - галантамина Вен-Чунг Ши и Джон А. Карлсон J. Org. Chem.; 1994; 59 (18) pp 5463–5465; Дои:10.1021 / jo00097a060
- ^ Кодама, Сумиаки; Хамасима, Йошио; Нишиде, Киёхару; Узел, Манабу (2004). «Полный синтез (-) - галантамина удаленной асимметричной индукцией». Angewandte Chemie International Edition. 43 (20): 2659–2661. Дои:10.1002 / anie.200353636.
- ^ а б Сатчароен, Вачирапорн; Маклин, Невилл Дж .; Кемп, Стивен С .; Лагерь, Николай П .; Браун, Ричард С. Д. (2007). «Стереоконтролируемый синтез (-) - галантамина». Органические буквы. 9 (10): 1867–1869. Дои:10.1021 / ol070255i. PMID 17429978.
- ^ Магнус, Филипп; Вменяемый, Нирадж; Fauber, Benjamin P .; Линч, Винс (2009). «Краткий синтез (-) - галантамина и (±) -кодеина посредством внутримолекулярного алкилирования производного фенола». Журнал Американского химического общества. 131 (44): 16045–16047. Дои:10.1021 / ja9085534. PMID 19835379.
- ^ Чида, Норитака; Като, Томоаки; Ямада, Хисако (2010). «Полный синтез (+) - и (-) - галантамина». Гетероциклы. 82: 563. Дои:10.3987 / COM-10-S (E) 27. Архивировано из оригинал на 22.07.2011.
- ^ Ху, Сян-Донг; Ту, Юн Цян; Чжан, Эн; Гао, Шуанху; Ван, Шаохуа; Ван, Эксия; Фань, Чун-Ан; Ван, Мин (2006). «Полный синтез (±) -галантамина ‡». Органические буквы. 8 (9): 1823–1825. Дои:10.1021 / ol060339b. PMID 16623560.
- ^ Исикава, Терухико; Кудо, Казухиро; Куроябу, Кен; Учида, Сатоши; Кудо, Такаяки; Сайто, Сейки (2008). "Двойные домино циклизации Майкла-Клейзена: мощный общий инструмент для введения четвертичных стереоцентров в C (4) циклогексан-1,3-дионов и полного синтеза различных семейств стерически конгестированных алкалоидов". Журнал органической химии. 73 (19): 7498–7508. Дои:10.1021 / jo801316s. PMID 18781800.
- ^ Разработка экспериментального масштабного процесса для лекарственного средства против болезни Альцгеймера (-) - галантамина с использованием крупномасштабного фенольного окислительного взаимодействия и индуцированного кристаллизацией хирального превращения Бернхард Кюенбург, Ласло Чолльнер, Йоханнес Фрёлих и Ульрих Джордис Org. Процесс Res. Dev .; 1999; 3 (6) pp 425–431; (Статья) Дои:10.1021 / op990019q
- ^ Дивергентный энантиоселективный синтез (-) - галантамина и (-) - морфина Барри М. Трост, Вэйпин Тан и Ф. Дин Тосте Варенье. Chem. Soc.; 2005; 127 (42) стр. 14785–14803; (Статья) Дои:10.1021 / ja054449 +
- ^ а бром, ацетат натрия, уксусная кислота, утюг, rt б карбонат калия, 2 дня c Troc-Cl, DMAP, Пиридин, дихлорметан d палладий, Лиганд Троста, триэтиламин, дихлорметан е 1,5% мол. ЦОХ, CH (OMe)3, метанол ж ДИБАЛ-Н, толуол, −78 ° C, 1 час грамм трифенилфосфин, ацетонциангидрин, ДИАДА, диэтиловый эфир час 2,20 мол.% ЦОН, THF, воды я 15 мол.% Ацетат палладия (II), 15 мол.% dppp, 3 экв. Ag2CO3, толуол, 107 ° С j диоксид селена динатрий фосфат диоксан, 150 ° C 3 часа k метиламин, метанол л 4 экв. ДИБАЛ-Н, м водный NaH2PO4 п NaCNBH3