Кинетический изотопный эффект - Kinetic isotope effect
![{ displaystyle { begin {matrix} { ce {{CN ^ {-}} + {^ {12} CH3-Br} -> [k_ {12}] {^ {12} CH3-CN} + Br ^ {-}}} { ce {{CN ^ {-}} + {^ {13} CH3-Br} -> [k_ {13}] {^ {13} CH3-CN} + Br ^ {-}}} {} end {matrix}} qquad { text {KIE}} = { frac {k_ {12}} {k_ {13}}} = 1.082 pm 0.008}](https://wikimedia.org/api/rest_v1/media/math/render/svg/438109ea220fd190ccc57f3e2c3726c47c24aae0)
В реакции бромистый метил с цианид,
кинетический изотопный эффект углерода в метильная группа оказалось равным 1,082 ± 0,008.[1][2]
В физическая органическая химия, а кинетический изотопный эффект (KIE) - изменение скорость реакции из химическая реакция когда один из атомы в реагенты заменяется одним из изотопы.[3] Формально это соотношение константы скорости для реакций с участием света (kL) и тяжелый (kЧАС) изотопно замещенные реагенты (изотопологи):

Это изменение скорости реакции является квантово-механическим эффектом, который в первую очередь является результатом более тяжелого изотопологи имея более низкий колебательный частоты по сравнению с их более легкими аналогами. В большинстве случаев это означает, что более тяжелым изотопологам требуется больше энергии для достижения переходное состояние (или, в редких случаях, предел диссоциации ) и, как следствие, более медленная скорость реакции. Изучение кинетических изотопных эффектов может помочь в выяснении механизм реакции определенных химических реакций и иногда используется при разработке лекарств для улучшения неблагоприятных фармакокинетика за счет защиты метаболически уязвимых связей C-H.
Фон
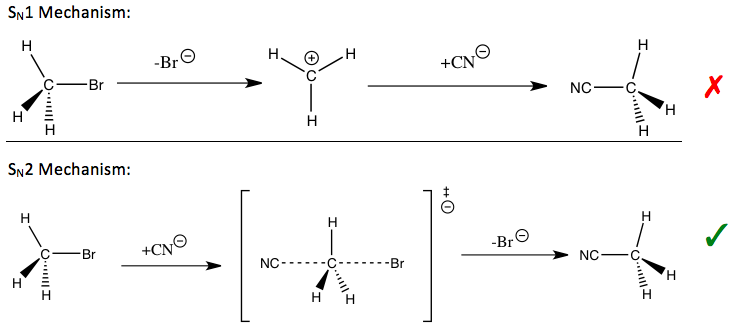
Кинетический изотопный эффект считается одним из наиболее важных и чувствительных инструментов для изучения механизмы реакции, знание которых позволяет улучшить желательные качества соответствующих реакций. Например, кинетические изотопные эффекты могут быть использованы для определения того, нуклеофильное замещение реакция следует за мономолекулярный (SN1) или бимолекулярный (SN2) путь.
В реакции бромистый метил и цианид (показано во введении), наблюдаемый кинетический изотопный эффект метильного углерода указывает на SN2 механизм.[1] В зависимости от пути могут использоваться разные стратегии для стабилизации переходное состояние из этап определения ставки реакции и улучшить скорость реакции и избирательность, что важно для промышленного применения.
Изотопные изменения скорости наиболее выражены, когда относительная масса изменение наиболее велико, поскольку эффект связан с частотами колебаний затронутых связей. Например, изменение водород атом (H) на его изотоп дейтерий (D) означает увеличение массы на 100%, тогда как при замене углерод -12 с углеродом-13 масса увеличивается всего на 8 процентов. Скорость реакции с участием связи C – H обычно в 6–10 раз выше, чем скорость соответствующей связи C – D, тогда как скорость реакции 12C реакция всего на 4 процента быстрее, чем соответствующая 13C реакция[4]:445 (хотя в обоих случаях изотоп один атомная единица массы тяжелее).
Изотопное замещение может изменять скорость реакции множеством способов. Во многих случаях разницу в скорости можно объяснить, отметив, что масса атома влияет на частота колебаний из химическая связь что он образует, даже если поверхность потенциальной энергии ибо реакция почти идентична. Более тяжелые изотопы будут (классически ) приводят к более низким частотам вибрации, или, если смотреть квантово-механически, будет ниже энергия нулевой точки. При более низкой энергии нулевой точки необходимо подавать больше энергии для разрыва связи, что приводит к более высокому энергия активации для разрыва связи, что, в свою очередь, снижает измеряемую скорость (см., например, Уравнение Аррениуса ).[3][4]:427
Классификация
Первичные кинетические изотопные эффекты
А первичный кинетический изотопный эффект может быть обнаружен при образовании или разрыве связи с меченным изотопом атомом.[3][4]:427 В зависимости от способа исследования кинетического изотопного эффекта (параллельное измерение скоростей против. межмолекулярная конкуренция против. внутримолекулярная конкуренция), наблюдение первичного кинетического изотопного эффекта указывает на разрыв / образование связи с изотопом на стадии ограничения скорости или последующей стадии (стадиях) определения продукта. (Заблуждение, что первичный кинетический изотопный эффект должен отражать разрыв / образование связи с изотопом на стадии ограничения скорости, часто повторяется в учебниках и первичной литературе: см. раздел о эксперименты ниже.)[5]
Для ранее упомянутых реакций нуклеофильного замещения первичные кинетические изотопные эффекты были исследованы как для уходящих групп, так и для нуклеофилов, и для α-углерода, на котором происходит замещение. Интерпретация кинетических изотопных эффектов уходящей группы поначалу была затруднена из-за значительного вклада температурно-независимых факторов. Кинетические изотопные эффекты на α-углероде могут быть использованы для понимания симметрии переходного состояния в SN2, хотя этот кинетический изотопный эффект менее чувствителен, чем тот, который был бы идеальным, также из-за вклада не колебательных факторов.[1]
Вторичные кинетические изотопные эффекты
А вторичный кинетический изотопный эффект наблюдается, когда связь с меченным изотопом атомом в реагенте не разорвана или не образована.[3][4]:427 Вторичные кинетические изотопные эффекты обычно намного меньше, чем первичные кинетические изотопные эффекты; однако вторичные изотопные эффекты дейтерия могут достигать 1,4 на атом дейтерия, и были разработаны методы для измерения изотопных эффектов тяжелых элементов с очень высокой точностью, поэтому вторичные кинетические изотопные эффекты все еще очень полезны для выяснения механизмов реакции.
Для вышеупомянутых реакций нуклеофильного замещения вторичные кинетические изотопные эффекты водорода у α-углерода обеспечивают прямые средства различения между SN1 и SN2 реакции. Было обнаружено, что SN1 реакции обычно приводят к большим вторичным кинетическим изотопным эффектам, приближающимся к их теоретическому максимуму около 1,22, в то время как SN2 реакции обычно приводят к первичным кинетическим изотопным эффектам, которые очень близки или меньше единицы. Кинетические изотопные эффекты, превышающие 1, называются нормальные кинетические изотопные эффекты, а кинетические изотопные эффекты, меньшие единицы, называются обратные кинетические изотопные эффекты. В общем, ожидается, что меньшие силовые постоянные в переходном состоянии приведут к нормальному кинетическому изотопическому эффекту, а большие силовые постоянные в переходном состоянии, как ожидается, приведут к обратному кинетическому изотопическому эффекту, когда вклады валентных колебаний доминируют над кинетическим изотопическим эффектом.[1]
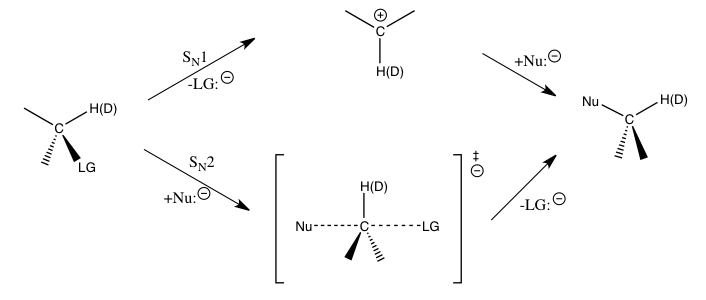
Величины таких вторичных изотопных эффектов на α-углероде в значительной степени определяются Cα-H (D) колебания. Для SN1, поскольку углерод превращается в пр.2 гибридизированный ион карбения во время переходного состояния для стадии, определяющей скорость, с увеличением Cα-H (D), обратный кинетический изотопный эффект можно было бы ожидать, если бы были важны только валентные колебания. Обнаружено, что наблюдаемые большие нормальные кинетические изотопные эффекты вызываются значительными внеплоскостными изгибными колебательными колебаниями при переходе от реагентов к переходному состоянию образования карбения. Для SN2, деформационные колебания по-прежнему играют важную роль для кинетического изотопного эффекта, но вклады валентных колебаний более сопоставимы по величине, и результирующий кинетический изотопный эффект может быть нормальным или обратным в зависимости от конкретных вкладов соответствующих колебаний.[1][6][7]
Теория
Теоретическое рассмотрение изотопных эффектов в значительной степени опирается на теория переходного состояния, который предполагает единую поверхность потенциальной энергии для реакции и барьер между реагентами и продуктами на этой поверхности, на вершине которой находится переходное состояние.[8][9] Кинетический изотопный эффект возникает в основном из-за изменений основных колебательных состояний, вызванных изотопическим возмущением вдоль пути минимальной энергии поверхности потенциальной энергии, что может быть объяснено только с помощью квантово-механических обработок системы. В зависимости от массы атома, движущегося по координате реакции, и характера (ширины и высоты) энергетического барьера, квантово-механическое туннелирование также может вносить большой вклад в наблюдаемый кинетический изотопический эффект и, возможно, требует отдельного рассмотрения в дополнение к «полуклассической» модели теории переходного состояния.[8]
Кинетический изотопный эффект дейтерия (2H KIE), безусловно, является наиболее распространенным, полезным и хорошо изученным типом кинетического изотопного эффекта. Точное предсказание численного значения кинетического изотопного эффекта дейтерия с использованием расчетов по теории функционала плотности в настоящее время является относительно обычным делом. Более того, несколько качественных и полуколичественных моделей позволяют делать грубые оценки изотопных эффектов дейтерия без расчетов, часто предоставляя достаточно информации для рационализации экспериментальных данных или даже для подтверждения или опровержения различных механистических возможностей. Исходные материалы, содержащие дейтерий, часто коммерчески доступны, что делает синтез исходных материалов, обогащенных изотопами, относительно простым. Кроме того, из-за большой относительной разницы в массе дейтерия и протия и сопутствующих различий в частотах колебаний, величина изотопного эффекта больше, чем у любой другой пары изотопов, кроме протия и трития,[10] позволяет легко измерить и интерпретировать как первичные, так и вторичные изотопные эффекты. Напротив, вторичные эффекты обычно очень малы для более тяжелых элементов и близки по величине к экспериментальной неопределенности, что усложняет их интерпретацию и ограничивает их полезность. В контексте изотопных эффектов водород часто используется для обозначения легкого изотопа протия (1H), в частности. В остальной части статьи ссылка на водород и дейтерий параллельные грамматические конструкции или прямые сравнения между ними следует интерпретировать как относящиеся к протию и дейтерию.[11]
Теория кинетических изотопных эффектов была впервые сформулирована Джейкоб Бигелейзен в 1949 г.[12][4]:427 Общая формула Бигелейзена для кинетических изотопных эффектов дейтерия (которая также применима к более тяжелым элементам) приводится ниже. Он использует теорию переходного состояния и статистическую механическую обработку поступательных, вращательных и колебательных уровней для расчета констант скорости. kЧАС и kD. Однако эта формула является «полуклассической», поскольку в ней не учитывается вклад квантового туннелирования, который часто вводится как отдельный поправочный коэффициент. Формула Бигелейзена также не рассматривает различия в несвязанных отталкивающих взаимодействиях, вызванных немного более короткой связью C – D по сравнению со связью C – H. В уравнении величины с нижними индексами H или D относятся к замещенным водородом или дейтерием частицам соответственно, тогда как величины с или без двойного крестика ‡ относятся к переходному состоянию или основному состоянию реагента соответственно.[7][13] (Строго говоря, член, возникающий из-за изотопной разницы в коэффициентах передачи, также должен быть включен.[14])

- ,
![{ displaystyle { frac {k _ {{ ce {H}}}} {k _ {{ ce {D}}}}} = left ({ frac { sigma _ {{ ce {H}}) } sigma _ {{ ce {D}}} ^ { ddagger}} { sigma _ {{ ce {D}}} sigma _ {{ ce {H}}} ^ { ddagger}} } right) left ({ frac {M _ {{ ce {H}}} ^ { ddagger} M _ {{ ce {D}}}}} {M _ {{ ce {D}}} ^ { ddagger} M _ {{ ce {H}}}} right) ^ { frac {3} {2}} left ({ frac {I_ {x { ce {H}}} ^ { ddagger} I_ {y { ce {H}}} ^ { ddagger} I_ {z { ce {H}}} ^ { ddagger}} {I_ {x { ce {D}}} ^ { ddagger} I_ {y { ce {D}}} ^ { ddagger} I_ {z { ce {D}}} ^ { ddagger}}} { frac {I_ {x { ce {D}} } I_ {y { ce {D}}} I_ {z { ce {D}}}} {I_ {x { ce {H}}} I_ {y { ce {H}}} I_ {z { ce {H}}}}} right) ^ { frac {1} {2}} left ({ frac { prod limits _ {i = 1} ^ {3N ^ { ddagger} - 7} { frac {1-e ^ {- u_ {i { ce {D}}} ^ { ddagger}}} {1-e ^ {- u_ {i { ce {H}}} ^ { ddagger}}}}} { prod limits _ {i = 1} ^ {3N-6} { frac {1-e ^ {- u_ {i { ce {D}}}}} {1- e ^ {- u_ {i { ce {H}}}}}}}} right) e ^ {- { frac {1} {2}} left [ sum limits _ {i = 1} ^ {3N ^ { ddagger} -7} (u_ {i { ce {H}}} ^ { ddagger} -u_ {i { ce {D}}} ^ { ddagger}) - sum пределы _ {i = 1} ^ {3N-6} (u_ {i { ce {H}}} - u_ {i { ce {D}}}) right]}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/93f26faede9d0fba35d6f675e641c716e7284c0d)
где мы определяем
- и .


Здесь, час это Постоянная Планка, kB это Постоянная Больцмана, частота вибрации, выраженная в волновые числа, c это скорость света, NА это Константа Авогадро, и р это универсальная газовая постоянная. ΣИкс (X = H или D) - числа симметрии для реагентов и переходных состояний. В MИкс - молекулярные массы соответствующих частиц, а яqИкс (q = Икс, у, или же z) члены представляют собой моменты инерции относительно трех главных осей. В тыяИкс прямо пропорциональны соответствующим частотам колебаний, νя, а колебательные энергия нулевой точки (Смотри ниже). Целые числа N и N‡ - количество атомов в реагентах и переходных состояниях соответственно.[7] Приведенное выше сложное выражение можно представить как произведение четырех отдельных факторов:[7]

- .

Для частного случая изотопных эффектов дейтерия мы будем утверждать, что первые три члена можно рассматривать как равные или хорошо приближенные к единице. Первый фактор S (содержащий σИкс) - это отношение чисел симметрии для различных видов. Это будет рациональное число (отношение целых чисел), которое зависит от количества вращений молекул и связей, ведущих к перестановке идентичных атомов или групп в реагентах и переходном состоянии.[13] Для систем с низкой симметрией все σИкс (реагент и переходное состояние) будут равны единице; таким образом S часто можно пренебречь. В MMI фактор (содержащий MИкс и яqИкс) относится к соотношению молекулярных масс и моментов инерции. Поскольку водород и дейтерий имеют тенденцию быть намного легче по сравнению с большинством реагентов и переходных состояний, существует небольшая разница в молекулярных массах и моментах инерции между молекулами, содержащими H и D, поэтому MMI Фактор обычно также приближается к единице. В EXC фактор (содержащий произведение колебательного функции раздела ) корректирует кинетический изотопический эффект, вызванный реакциями колебательно-возбужденных молекул. Доля молекул с достаточной энергией, чтобы иметь колебания связи A – H / D в возбужденном состоянии, обычно мала для реакций при комнатной температуре или около нее (связи с водородом обычно колеблются на расстоянии 1000 см−1 или выше, поэтому exp (-тыя) = ехр (-hνя/kBТ) <0,01 при 298 K, что приводит к незначительному вкладу 1 – exp (-тыя) факторы). Следовательно, для кинетических изотопных эффектов водорода / дейтерия в наблюдаемых значениях обычно преобладает последний фактор, ZPE (экспоненциальная функция разностей нулевой энергии колебаний), состоящая из вкладов разностей нулевой энергии для каждой из колебательных мод реагентов и переходного состояния, которые можно представить следующим образом:[7]
- ,
![{ displaystyle { begin {align} { frac {k _ {{ ce {H}}}} {k _ {{ ce {D}}}}} & cong exp left {- { frac {1} {2}} left [ sum limits _ {i = 1} ^ {3N ^ { ddagger} -7} (u_ {i { ce {H}}} ^ { ddagger} -u_ {i { ce {D}}} ^ { ddagger}) - sum limits _ {i = 1} ^ {3N-6} (u_ {i { ce {H}}} - u_ {i { ce {D}}}) right] right } & cong exp left [ sum _ {i} ^ { mathrm {(реагировать.)}} { frac {1} {2 }} Delta u_ {i} - sum _ {i} ^ { mathrm {(TS)}} { frac {1} {2}} Delta u_ {i} ^ { ddagger} right] конец {выровнен}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/20d669d624e10fcb89d116442c5543998ea4fbf8)
где мы определяем
- и .


Суммы в показателе степени второго выражения можно интерпретировать как пробегающие все колебательные моды основного состояния реагента и переходного состояния. В качестве альтернативы их можно интерпретировать как переход по тем режимам, уникальным для реагента или переходного состояния, или чьи частоты колебаний существенно изменяются при продвижении по координате реакции. Остальные пары колебательных мод реагента и переходного состояния очень похожи. и , и сокращения происходят при вычислении сумм в экспоненте. Таким образом, на практике дейтериевые КИЭ часто в значительной степени зависят от нескольких ключевых форм колебаний из-за этого подавления, что позволяет проводить качественный анализ kЧАС/kD возможный.[13]


Как уже упоминалось, особенно для замещения водород / дейтерий, большинство кинетических изотопных эффектов возникает из-за разницы в энергия нулевой точки (ZPE) между реагентами и переходным состоянием рассматриваемых изотопологов, и это различие можно понять качественно с помощью следующего описания: в пределах Приближение Борна – Оппенгеймера, поверхность потенциальной энергии одинакова для обоих изотопов. Однако квантово-механическое рассмотрение энергии вводит дискретные колебательные уровни на эту кривую, и самое низкое возможное энергетическое состояние молекулы соответствует самому низкому уровню колебательной энергии, который немного выше по энергии, чем минимум кривой потенциальной энергии. Эта разница, называемая нулевой энергией, является проявлением принципа неопределенности Гейзенберга, который требует неопределенности в длине связи C-H или C-D. Поскольку более тяжелые (в данном случае дейтерированные) частицы ведут себя более «классически», их уровни колебательной энергии ближе к классической кривой потенциальной энергии, и она имеет более низкую нулевую энергию. Разница в нулевой энергии между двумя изотопными частицами, по крайней мере, в большинстве случаев, уменьшается в переходном состоянии, поскольку константа силы связи уменьшается во время разрыва связи. Следовательно, более низкая энергия нулевой точки дейтерированных частиц переводится в большую энергию активации его реакции, как показано на следующем рисунке, что приводит к нормальному кинетическому изотопному эффекту.[15] Этот эффект, в принципе, следует учитывать все 3N−6 мод колебаний исходного материала и 3N‡−7 мод колебаний в переходном состоянии (одна мода, соответствующая координате реакции, отсутствует в переходном состоянии, поскольку связь разрывается и нет восстанавливающей силы против движения). В гармонический осциллятор является хорошим приближением для колеблющейся связи, по крайней мере, для низкоэнергетических колебательных состояний. Квантовая механика дает колебательную нулевую энергию как . Таким образом, мы можем легко интерпретировать множитель ½ и суммы члены по колебательным модам в основном и переходном состоянии в показателе степени упрощенной формулы выше. Для гармонического осциллятора частота колебаний обратно пропорциональна квадратному корню из приведенной массы колебательной системы:


- ,

куда kж это силовая постоянная. Более того, приведенная масса аппроксимируется массой легкого атома системы, X = H или D. Поскольку мD примерно 2мЧАС,
- .

В случае гомолитической диссоциации связи C – H / D член переходного состояния исчезает, и, если пренебречь другими колебательными модами, kЧАС/kD = ехр (½Δтыя). Таким образом, больший изотопный эффект наблюдается для более жесткой («более сильной») связи C – H / D. Для большинства представляющих интерес реакций атом водорода переносится между двумя атомами с переходным состоянием [A ··· H ··· B]‡ и колебательные моды в переходном состоянии необходимо учитывать. Тем не менее, по-прежнему верно, что разрыв связи с более высокой частотой колебаний даст больший изотопный эффект.

Чтобы рассчитать максимально возможное значение для нетуннелирующего дейтериевого КИЭ, мы рассмотрим случай, когда разность нулевой энергии между валентными колебаниями типичной углерод-водородной связи (3000 см−1) и углерод-дейтерийная связь (2200 см−1) исчезает в переходном состоянии (разность энергий (1/2) (3000 - 2200 см−1) = 400 см−1, или около 1,15 ккал / моль), без какой-либо компенсации из нулевой разности энергий в переходном состоянии (например, из симметричного участка A ··· H ··· B, который является уникальным для переходного состояния). Упрощенная формула, приведенная выше, предсказывает максимум для kЧАС/kD как 6.9. Если также учитывать полное исчезновение двух изгибных колебаний, kЧАС/kD могут быть предсказаны такие большие значения, как 15-20. Однако маловероятно, что изгибные частоты исчезнут в переходном состоянии, и есть лишь несколько случаев, когда kЧАС/kD значения превышают 7-8 при комнатной температуре. Более того, часто обнаруживается, что туннелирование является основным фактором, когда они превышают такие значения. Ценность kЧАС/kD ~ 10 считается максимальным для полуклассического первичного кинетического изотопного эффекта (без туннелирования) для реакций, протекающих при температуре около 298 К. (Формула для kЧАС/kD имеет температурную зависимость, поэтому при более низких температурах возможны более значительные изотопные эффекты).[16] В зависимости от природы переходного состояния переноса водорода (симметричный по сравнению с «ранним» или «поздним» и линейный по сравнению с изогнутым), степень приближения эффекта первичного изотопа дейтерия к этому максимуму варьируется. Модель, разработанная Westheimer предсказал, что симметричный (термонейтральный, Постулат Хаммонда ), линейные переходные состояния имеют наибольшие изотопные эффекты, в то время как переходные состояния, которые являются "ранними" или "поздними" (для экзотермических или эндотермических реакций, соответственно) или нелинейными (например, циклическими), демонстрируют меньшие эффекты. С тех пор эти предсказания получили широкую экспериментальную поддержку.[17]
Для вторичных изотопных эффектов дейтерия Streitwieser предположил, что ослабление (или усиление, в случае обратного изотопического эффекта) изотопных мод из основного состояния реагента в переходное состояние в значительной степени ответственны за наблюдаемые изотопические эффекты. Эти изменения объясняются изменением стерического окружения, когда углерод, связанный с H / D, подвергается регибридизации из sp.3 к sp2 или наоборот (вторичный кинетический изотопный эффект α), или ослабление связи из-за гиперконъюгации в случаях, когда карбокатион образуется на расстоянии одного атома углерода (вторичный кинетический изотопный эффект β). Теоретический максимум этих изотопных эффектов составляет kЧАС/kD = 20.5 ≈ 1,4. Для вторичного кинетического изотопного эффекта в α-положении регибридизация из sp3 к sp2 производит нормальный изотопный эффект, а регибридизация из sp2 к sp3 приводит к обратному изотопному эффекту с теоретическим минимумом kЧАС/kD = 2-0.5 ≈ 0,7. На практике, kЧАС/kD ~ 1.1-1.2 и kЧАС/ kD ~ 0.8-0.9 характерны для α вторичных кинетических изотопных эффектов, а kЧАС/kD ~ 1,15–1,3 характерны для β вторичного кинетического изотопного эффекта. Для реагентов, содержащих несколько изотопно-замещенных β-атомов водорода, наблюдаемый изотопный эффект часто является результатом согласованного действия нескольких H / D в β-положении. В этих случаях действие каждого изотопно-меченого атома мультипликативно, а в случаях, когда kЧАС/kD > 2 не редкость.[18]
Следующие простые выражения, связывающие кинетические изотопные эффекты дейтерия и трития, также известные как Уравнение Суэйна (или уравнения Суэйна-Шада-Стиверса), могут быть получены из общего выражения, приведенного выше, с использованием некоторых упрощений:[8][19]
- ;

т.е.
- .

При выводе этих выражений использовалось разумное приближение, согласно которому приведенные массы приблизительно равны массам водорода, дейтерия или трития. Кроме того, предполагалось, что колебательное движение аппроксимируется гармоническим осциллятором, так что (X = H, D или T). Нижний индекс "s"относится к этим" полуклассическим "кинетическим изотопным эффектам, которые не учитывают квантовое туннелирование. Вклады туннелирования следует рассматривать отдельно как поправочный коэффициент.

Для изотопных эффектов с участием элементов, отличных от водорода, многие из этих упрощений недействительны, и величина изотопного эффекта может сильно зависеть от некоторых или всех игнорируемых факторов. Таким образом, кинетические изотопные эффекты для элементов, отличных от водорода, часто гораздо труднее рационализировать или интерпретировать. Во многих случаях, особенно в реакциях переноса водорода, вклад в кинетические изотопные эффекты от туннелирования значительный (см. Ниже).
Туннелирование
В некоторых случаях наблюдается дополнительное увеличение скорости для более легкого изотопа, возможно, из-за квантово-механическое туннелирование. Обычно это наблюдается только для реакций, связанных с атомами водорода. Туннелирование происходит, когда молекула проникает через потенциальный энергетический барьер, а не через него.[20][21] Хотя это не разрешено законами классическая механика, частицы могут проходить через классически запрещенные области пространства в квантовой механике, основанной на дуальность волна-частица.[22]
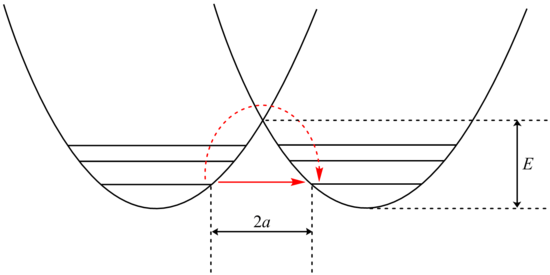
Анализ туннелирования может быть выполнен с использованием модификации Белла Уравнение Аррениуса, который включает добавление коэффициента туннелирования Q:

где A - параметр Аррениуса, E - высота барьера и

куда и


Рассмотрение β Член показывает экспоненциальную зависимость от массы частицы. В результате туннелирование более вероятно для более легкой частицы, такой как водород. Просто удвоив массу туннельного протона, заменив его его дейтерий изотоп резко снижает скорость таких реакций. В результате наблюдаются очень большие кинетические изотопные эффекты, которые нельзя объяснить различиями в энергиях нулевой точки.

В дополнение β член линейно зависит от ширины барьера, 2а. Как и в случае с массой, туннелирование лучше всего при небольшой ширине барьера. Оптимальное туннельное расстояние протонов между донорным и акцепторным атомами составляет 0,4 Å.[24]
Туннелирование квантово-механический эффект, связанный с законами волновой механики, а не кинетика. Следовательно, туннелирование имеет тенденцию становиться более важным при низких температурах, когда даже самые маленькие барьеры кинетической энергии не могут быть преодолены, но могут быть преодолены.[20]
Петр С. Зуев и др. Сообщенные константы скорости расширения цикла 1-метилциклобутилфторкарбена составляют 4,0 × 10−6/ с в азоте и 4,0 x 10−5/ с в аргоне при 8 кельвинах. Они подсчитали, что при температуре 8 кельвинов реакция будет протекать через одно квантовое состояние реагента, так что указанная константа скорости не зависит от температуры, а вклад туннелирования в скорость был на 152 порядка величины больше, чем вклад перехода через переходное состояние. энергетический барьер.[25]
Таким образом, несмотря на то, что обычные химические реакции имеют тенденцию резко замедляться при понижении температуры, туннельные реакции редко меняются вообще. Частицы, которые проходят сквозь активационный барьер, являются прямым результатом того факта, что волновая функция промежуточных частиц, реагента или продукта не ограничивается энергетической ямой конкретного желоба вдоль энергетической поверхности реакции, но может «просачиваться». в следующий минимум энергии. В свете этого туннелирование должен не зависеть от температуры.[20][3]
Для отрыва водорода от газообразных н-алканов и циклоалканов атомами водорода в интервале температур 363–463 K данные кинетического изотопного эффекта H / D характеризовались небольшими предэкспоненциальный коэффициент соотношения АЧАС/АD от 0,43 до 0,54 и большие различия в энергии активации от 9,0 до 9,7 кДж / моль. Основывая свои аргументы на теория переходного состояния, маленький А соотношения факторов, связанные с большой разницей в энергии активации (обычно около 4,5 кДж / моль для связей C – H (D)), предоставили убедительные доказательства туннелирования. Для целей этого обсуждения важно, чтобы А соотношение факторов для различных используемых парафинов было примерно постоянным во всем температурном диапазоне.[26]
Наблюдение за тем, что туннелирование не полностью зависит от температуры, можно объяснить тем фактом, что не все молекулы определенного вида занимают свое основное колебательное состояние при различных температурах. Добавление тепловой энергии к потенциальной энергетической яме может вызвать заселение более высоких колебательных уровней, отличных от основного состояния. Для обычной кинетической реакции это возбуждение будет иметь лишь небольшое влияние на скорость. Однако для туннельной реакции разница между энергия нулевой точки и первый уровень вибрационной энергии мог быть огромным. Член поправки на туннелирование Q линейно зависит от ширины барьера, и эта ширина значительно уменьшается по мере увеличения числа колебательные режимы на Потенциал Морзе увеличивать. Уменьшение ширины барьера может иметь такое огромное влияние на скорость туннелирования, что даже небольшая популяция возбужденных колебательных состояний будет доминировать в этом процессе.[20][3]Чтобы определить, участвует ли туннелирование в KIE реакции с H или D, рассматриваются несколько критериев:
- Δ (EаЧАС-EаD)> Δ (ZPEЧАС-ZPED) (Eа= энергия активации; ZPE = энергия нулевой точки)
- Реакция по-прежнему протекает при более низких температурах.
- В Аррениус предэкспоненциальные факторы АD/АЧАС не равно 1.
- Большой негатив энтропия активации.
- Геометрии реагентов и продуктов обычно очень похожи.[20]
Также для реакций, в которых изотопы включают H, D и T, критерием туннелирования являются соотношения Суэйна-Шаада, которые сравнивают константы скорости (k) реакций, в которых происходит обмен H, D или T:
- kЧАС/kТ=(kD/kТ)Икс и kЧАС/kТ=(kЧАС/kD)Y
В органических реакциях этот эффект туннелирования протонов наблюдался в таких реакциях, как депротонирование и йодирование нитропропан с затрудненным пиридин основание[27] с сообщенным KIE 25 при 25 ° C:

и в 1,5-сигматропный водородный сдвиг[28] хотя наблюдается, что трудно экстраполировать экспериментальные значения, полученные при повышенных температурах, на более низкие температуры:[29][30]

Долгое время предполагалось, что высокая эффективность ферментативного катализа в реакциях переноса протона или гидрид-иона может быть частично связана с квантово-механическим туннельным эффектом. Окружающая среда в активном центре фермента размещает донорный и акцепторный атомы близко к оптимальному расстоянию туннелирования, где боковые цепи аминокислот могут «заставить» донорный и акцепторный атом сблизиться друг с другом за счет электростатических и нековалентных взаимодействий. Также возможно, что фермент и его необычная гидрофобная среда внутри реакционного участка обеспечивают вибрацию, способствующую туннелированию.[31] Исследования кетостероидизомеразы предоставили экспериментальные доказательства того, что фермент на самом деле усиливает связанное движение / водородное туннелирование, сравнивая первичные и вторичные кинетические изотопные эффекты реакции в условиях, катализируемых ферментами, и неферментативных катализаторах.[32]
Существует множество примеров туннелирования протонов в реакциях, катализируемых ферментами, которые были обнаружены KIE. Хорошо изученным примером является метиламиндегидрогеназа, где большие первичные КИЭ 5–55 наблюдались на стадии переноса протона.[33]

Другим примером туннельного вклада в перенос протона в ферментативных реакциях является реакция, осуществляемая алкогольдегидрогеназа. Конкурентные KIE для стадии переноса водорода при 25 ° C дали 3,6 и 10,2 для первичного и вторичного KIE, соответственно.[34]
Переходный кинетический изотопный эффект
Изотопный эффект, выраженный уравнениями, приведенными выше, относится только к реакциям, которые можно описать с помощью кинетика первого порядка. Во всех случаях, когда это невозможно, переходные кинетические изотопные эффекты следует учитывать при использовании уравнений ГЕБИК и ГЕБИФ.[35]
Эксперименты
Симмонс и Хартвиг называют следующие три случая основными типами экспериментов с кинетическим изотопным эффектом, включающими функционализацию связи C-H:[5]
- А) KIE определяется из абсолютных скоростей двух параллельных реакций

В этом эксперименте константы скорости для нормального субстрата и его меченого изотопами аналога определяются независимо, и KIE получается как соотношение двух. Точность измеренного KIE сильно ограничена точностью, с которой может быть измерена каждая из этих констант скорости. Более того, воспроизведение точных условий в двух параллельных реакциях может быть очень сложной задачей. Тем не менее, измерение большого кинетического изотопного эффекта путем прямого сравнения констант скорости указывает на то, что разрыв связи C-H происходит на стадии, определяющей скорость. (Меньшее значение может указывать на изотопный эффект из-за предварительного равновесия, так что разрыв связи C-H происходит где-то до этапа, определяющего скорость.)
- Б) KIE определяется в результате межмолекулярного соревнования
В этом типе экспериментов используются те же субстраты, которые используются в эксперименте А, но они могут реагировать в одном и том же контейнере, а не в двух отдельных контейнерах. Кинетический изотопный эффект этого эксперимента определяется относительным количеством продуктов, образованных из C-H по сравнению с функционализацией C-D (или он может быть выведен из относительных количеств непрореагировавших исходных материалов). Чтобы наблюдать кинетический изотопный эффект, необходимо погасить реакцию до ее завершения (см. Раздел «Оценка» ниже). Обычно реакцию останавливают при низкой конверсии (от ~ 5 до 10% конверсии) или при использовании большого избытка (> 5 эквивалентов) смеси изотопов. Этот тип эксперимента гарантирует, что функционализация связей CH и CD происходит в абсолютно одинаковых условиях, а соотношение продуктов функционализации связей CH и CD может быть измерено с гораздо большей точностью, чем константы скорости в эксперименте A. Более того, только одно измерение концентрации продукта из одной пробы. Однако наблюдаемый кинетический изотопный эффект в этом эксперименте сложнее интерпретировать, поскольку он может означать, что разрыв связи C-H происходит во время стадии определения скорости или на стадии определения продукта, следующей за стадией определения скорости. Отсутствие кинетического изотопного эффекта, по крайней мере, в соответствии с Симмонсом и Хартвигом, тем не менее указывает на то, что разрыв связи C-H не происходит во время стадии, определяющей скорость.
- C) КИЭ определяется в результате внутримолекулярного соревнования
Этот тип эксперимента аналогичен эксперименту B, за исключением того, что на этот раз существует внутримолекулярная конкуренция за функционализацию связи C-H или C-D. В большинстве случаев субстрат имеет направляющую группу (DG) между связями C-H и C-D. Расчет кинетического изотопного эффекта в этом эксперименте и его интерпретация следуют тем же соображениям, что и в эксперименте B. Однако результаты экспериментов B и C будут отличаться, если в эксперименте B имеет место необратимое связывание изотопного субстрата. прежний к разрыву связи C-H или C-D. В таком сценарии изотопный эффект может наблюдаться в эксперименте C (где выбор изотопа может иметь место даже после связывания субстрата), но не в эксперименте B (поскольку выбор между расщеплением связи CH или CD уже сделан, как только субстрат связывается необратимо). В отличие от эксперимента B, реакцию не нужно останавливать при низком расходе изотопного исходного материала для получения точного kЧАС/kD, поскольку соотношение H и D в исходном материале составляет 1: 1, независимо от степени превращения.
Одним из примеров активации не-C-H различных изотопных эффектов, наблюдаемых в случае межмолекулярной (эксперимент B) и внутримолекулярной (эксперимент C) конкуренции, является фотолиз дифенилдиазометана в присутствии т-бутиламин. Чтобы объяснить этот результат, образование дифенилкарбена с последующей необратимой нуклеофильной атакой т-бутиламин. Поскольку существует небольшая изотопная разница в скорости нуклеофильной атаки, межмолекулярный эксперимент привел к KIE, близкому к 1. Однако во внутримолекулярном случае соотношение продуктов определяется переносом протона, который происходит после нуклеофильной атаки, процесс для который имеет существенный KIE 2.6.[36]

Таким образом, эксперименты A, B и C дадут результаты с разным уровнем точности и потребуют различных экспериментальных установок и способов анализа данных. В результате осуществимость каждого типа эксперимента будет зависеть от кинетического и стехиометрического профиля реакции, а также от физических характеристик реакционной смеси (например, гомогенный или гетерогенный). Более того, как отмечено в предыдущем абзаце, эксперименты предоставляют данные о кинетическом изотопном эффекте для различных стадий многостадийной реакции, в зависимости от относительного расположения стадии ограничения скорости, стадий определения продукта и / или расщепления CH / D. шаг.
Приведенные ниже гипотетические примеры иллюстрируют распространенные сценарии. Рассмотрим следующую диаграмму координат реакции. Для реакции с таким профилем все три эксперимента (A, B и C) дадут значительный первичный кинетический изотопный эффект:

С другой стороны, если реакция следует следующему энергетическому профилю, в котором разрыв связи CH или CD является необратимым, но происходит после стадии, определяющей скорость (RDS), в эксперименте A не будет наблюдаться значительного кинетического изотопного эффекта, поскольку общая скорость не зависит от изотопного замещения. Тем не менее, стадия необратимого разрыва связи C-H даст первичный кинетический изотопный эффект с двумя другими экспериментами, поскольку вторая стадия по-прежнему будет влиять на распределение продукта. Следовательно, с помощью экспериментов B и C можно наблюдать кинетический изотопный эффект, даже если разрыв связи C-H или C-D происходит не на этапе определения скорости, а на этапе определения продукта.

Большая часть кинетического изотопного эффекта возникает из-за колебательных разностей энергии нулевой точки между основным состоянием реагента и переходным состоянием, которые варьируются между реагентом и его изотопно замещенным аналогом. Хотя можно проводить сложные расчеты кинетических изотопных эффектов с использованием вычислительной химии, большая часть проделанной работы имеет более простой порядок, который включает исследование того, вызывают ли конкретные изотопные замещения обнаруживаемый кинетический изотопный эффект или нет. Колебательные изменения от изотопного замещения на атомах, удаленных от места, где происходит реакция, имеют тенденцию к нейтрализации между реагентом и переходным состоянием. Следовательно, наличие кинетического изотопного эффекта указывает на то, что меченный изотопом атом находится в центре реакции или очень близко к нему.
Отсутствие изотопного эффекта интерпретировать труднее: это может означать, что изотопно-меченый атом находится далеко от места реакции, но это также может означать, что существуют определенные компенсирующие эффекты, которые приводят к отсутствию наблюдаемого кинетического изотопного эффекта. Например, различия между реагентом и энергиями нулевой точки переходного состояния могут быть идентичными между нормальным реагентом и его версией, меченной изотопами. Альтернативно, это может означать, что изотопное замещение происходит в реакционном центре, но колебательные изменения, связанные со связями с этим атомом, происходят после стадии, определяющей скорость. Такой случай проиллюстрирован в следующем примере, в котором ABCD представляет собой атомный скелет молекулы.

Предполагая стационарные условия для промежуточного соединения ABC, общая скорость реакции будет следующей:
![{ frac {d [A]} {dt}} = { frac {k_ {1} k_ {3} [ABCD]} {k_ {2} [D] + k_ {3}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/f71c52221df559ae3b305086cf125e2cfbfa62c4)
Если первым шагом является определение скорости, это уравнение сводится к:
![{ frac {d [A]} {dt}} = k_ {1} [ABCD]](https://wikimedia.org/api/rest_v1/media/math/render/svg/6cfaea2beb4c13e320a49b4f66ede1c5a85d0fba)
Или, если вторым шагом является определение скорости, уравнение сводится к:
![{ frac {d [A]} {dt}} = { frac {k_ {1} k_ {3} [ABCD]} {k_ {2} [D]}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/ea50a8acf1a1307f4d9c2b4605f62578e2f79771)
В большинстве случаев изотопное замещение в A, особенно если это тяжелый атом, не изменит k1 или же k2, но, скорее всего, он изменит k3. Следовательно, если первый шаг является определением скорости, не будет наблюдаемого кинетического изотопного эффекта в общей реакции с изотопным мечением A, но он будет, если второй шаг будет определять скорость. Для промежуточных случаев, когда обе ступени имеют сопоставимые скорости, величина кинетического изотопного эффекта будет зависеть от соотношения k3 и k2.
Изотопное замещение D изменит k1 и k2 не затрагивая k3. Кинетический изотопный эффект всегда будет наблюдаться при такой замене, поскольку k1 появляется в упрощенном выражении скорости независимо от того, какой шаг определяет скорость, но он будет менее выражен, если второй шаг является определяющим из-за некоторой отмены между изотопными эффектами на k1 и k2. Этот результат связан с тем, что равновесные изотопные эффекты обычно меньше кинетических изотопных эффектов.
Изотопное замещение B явно изменит k3, но это также может изменить k1 в меньшей степени, если колебания связи B-C затронуты в переходном состоянии первой ступени. Таким образом, может иметь место небольшой изотопный эффект, даже если первым шагом является определение скорости.
Это гипотетическое соображение показывает, как наблюдение кинетических изотопных эффектов может быть использовано для исследования механизмов реакции. Существование кинетического изотопного эффекта указывает на изменение константы колебательной силы связи, связанной с изотопно-меченным атомом, на этапе регулирования скорости или до него. Сложные вычисления могут быть использованы для получения большого количества деталей о переходном состоянии из наблюдаемых кинетических изотопных эффектов. Однако чаще просто качественное знание того, что связь, связанная с изотопно меченным атомом, определенным образом изменена, может быть очень полезным.[37]Оценка соотношений констант скорости реакций межмолекулярной конкуренции
В реакциях конкуренции кинетический изотопный эффект рассчитывается по изотопному продукту или остающимся соотношениям реагентов после реакции, но эти соотношения сильно зависят от степени завершения реакции. Чаще всего изотопный субстрат состоит из молекул, помеченных в определенном положении, и их немеченых, обычных аналогов.[8] Также возможно в случае 13Кинетические изотопные эффекты C, а также аналогичные случаи, чтобы просто полагаться на естественное содержание изотопного углерода для экспериментов с кинетическим изотопным эффектом, устраняя необходимость изотопного мечения.[38] Два изотопных субстрата будут реагировать по одному и тому же механизму, но с разной скоростью. Соотношение между количествами двух компонентов в реагентах и продуктах, таким образом, будет постепенно изменяться в ходе реакции, и это постепенное изменение можно рассматривать следующим образом:[8]Предположим, что две изотопные молекулы A1 и А2, претерпевают необратимые реакции конкуренции следующим образом:
![{ displaystyle { begin {align} { ce {{A1} + {B} + {C} + cdots}} & { ce {-> [k_ {1}] P1}} { ce {{A2} + {B} + {C} + cdots}} & { ce {-> [k_ {2}] P2}} end {выровнено}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/8a3e1c66994bddc0e6b39e89f23e4e0ec7a47b5c)
Кинетический изотопный эффект для этого сценария равен:

Где F1 и F2 относятся к доле превращений для изотопных частиц A1 и А2, соответственно.
При этой обработке все остальные реагенты считаются неизотопными. Предполагая далее, что реакция имеет первый порядок по отношению к изотопному субстрату A, можно записать следующее общее выражение скорости для обеих этих реакций:
![{ displaystyle { text {rate}} = {- d [{ ce {A}} _ {n}] over dt} = k_ {n} times [{ ce {A}} _ {n} ] times f ([{ ce {B}}], [{ ce {C}}], cdots) { text {where}} n = 1 { text {или}} 2}](https://wikimedia.org/api/rest_v1/media/math/render/svg/26ca6ed80abd2295998fb23de5de165477f2848e)
Поскольку f ([B], [C],…) не зависит от изотопического состава A, его можно решить для обоих скоростных выражений с A1 и А2, и эти два могут быть приравнены для получения следующих соотношений:
![{ Displaystyle {1 над k_ {1}} times { ce {{ mathit {d}} [A1] over [A1]}} = {1 над k_ {2}} times { ce {{ mathit {d}} [A2] over [A2]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/ec95ce0a836f8db29e8a86da2d3a2390214f462c)
![{ displaystyle {1 over k_ {1}} times int limits _ { ce {[A1] ^ {0}}} ^ { ce {[A1]}} {d [{ ce {A }} '_ {1}] over [{ ce {A}}' _ {1}]} = {1 over k_ {2}} times int limits _ { ce {[A2] ^ {0}}} ^ { ce {[A2]}} {d [{ ce {A}} '_ {2}] over [{ ce {A}}' _ {2}]}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/8b3f5c1311b00398d60afa93ae27d58ac423fd32)
Где1]0 и [A2]0 - начальные концентрации A1 и А2, соответственно. Это приводит к следующему выражению кинетического изотопного эффекта:
![{ displaystyle {k_ {1} over k_ {2}} = { frac { ce { ln ([A1] / [A1] ^ {0})}} { ce { ln ([A2] / [A2] ^ {0})}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/a4f8b9f334d8799e9e6fc51d105e53d0148086f1)
Что также может быть выражено в единицах фракций конверсии двух реакций, F1 и F2, где 1-Fп= [Aп] / [Aп]0 для n = 1 или 2 следующим образом:


Что касается получения кинетических изотопных эффектов, смеси субстратов, содержащих стабильные изотопы, могут быть проанализированы с использованием масс-спектрометра, который дает соотношения изотопных молекул в исходном субстрате (определяемом здесь как [A2]0/ [A1]0= R0), в подложке после некоторого преобразования ([A2] / [A1] = R) или в продукте ([P2]/[П1] = Rп). Когда один из видов, например 2, является радиоактивным изотопом, его смесь с другими частицами также может быть проанализирована по его радиоактивности, которая измеряется в молярной активности, пропорциональной [A2]0 / ([A1]0+ [A2]0) ≈ [A2]0/ [A1]0 = R0 в исходной подложке [A2] / ([A1] + [A2]) ≈ [A2] / [A1] = R в подложке после некоторого преобразования и [R2] / ([Р1] + [R2]) ≈ [R2]/[Р1] = Rп, так что те же отношения, что и в другом случае, могут быть измерены, пока радиоактивный изотоп присутствует в индикаторных количествах. Такие соотношения также можно определить с помощью ЯМР-спектроскопии.[39][40]
При соблюдении состава субстрата следующее выражение кинетического изотопного эффекта в терминах R0 и R можно получить:
![{ displaystyle { text {KIE}} = { frac {k_ {1}} {k_ {2}}} = { frac { ln (1-F_ {1})} { ln [(1- F_ {1}) R / R_ {0}]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/9f819291ca2e6641dfaf1a2d31025c40927bdd7a)
Принимая соотношение R и R0 используя ранее полученное выражение для F2, получается:
![{ displaystyle {R over R_ {0}} = { ce {{ frac {[A2] / [A1]} {[A2] ^ 0 / [A1] ^ 0}}}} = { ce { { frac {[A2] / [A2] ^ 0} {[A1] / [A1] ^ 0}}}} = { frac {1-F_ {2}} {1-F_ {1}}} = (1-F_ {1}) ^ {(k_ {2} / k_ {1}) - 1}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/61529d236e7c75ed167c0a1e43f996134b54b908)
Изотопное обогащение исходного материала можно рассчитать по зависимости R / R0 на F1 для различных кинетических изотопных эффектов, что дает следующий рисунок. Из-за экспоненциальной зависимости даже очень низкие кинетические изотопные эффекты приводят к большим изменениям изотопного состава исходного материала при высоких конверсиях.

При отслеживании продуктов кинетический изотопный эффект можно рассчитать с использованием соотношения продуктов рп вместе с р0 следующее:
![{k_ {1} over k_ {2}} = { frac { ln (1-F_ {1})} { ln [1- (F_ {1} R_ {P} / R_ {0})] }}](https://wikimedia.org/api/rest_v1/media/math/render/svg/37f721659b133bfa69404286f90a8309fea92944)
Измерение кинетического изотопного эффекта при естественном изобилии
Измерение кинетического изотопного эффекта при естественном изобилии представляет собой простой общий метод измерения кинетических изотопных эффектов (KIE) для химические реакции выполнено с материалами природное изобилие. Этот метод измерения KIE преодолевает многие ограничения предыдущих методов измерения KIE. Измерения KIE из материалов, меченных изотопами, требуют нового синтеза для каждого материала, меченного изотопами (процесс часто чрезмерно сложен), реакции конкуренции и анализа.[5] Измерение KIE при природное изобилие позволяет избежать этих проблем, используя преимущества высокоточных количественных методов (спектроскопия ядерного магнитного резонанса, масс-спектрометрия изотопного отношения ) для выборочного измерения на сайте кинетическое фракционирование из изотопы, либо в продукте, либо в исходном материале для данного химическая реакция.
Одноимпульсный ЯМР
Количественный одиночный импульс спектроскопия ядерного магнитного резонанса (ЯМР) - это метод, пригодный для измерения кинетическое фракционирование из изотопы для измерений KIE естественного обилия. Паскаль и другие. были вдохновлены исследованиями, демонстрирующими резкие вариации содержания дейтерия в идентичных соединениях из разных источников, и выдвинули гипотезу о том, что ЯМР можно использовать для измерения кинетических изотопных эффектов дейтерия в естественных условиях.[41][42] Паскаль и его коллеги проверили свою гипотезу, изучив реакция вставки диметилдиазомалоната в циклогексан. Паскаль и др. измерил KIE 2,2, используя 2
ЧАС
ЯМР для материалов с естественным изобилием.[42]

Синглтон и его коллеги продемонстрировали возможности 13
C
Измерения KIE естественного обилия на основе ЯМР для изучения механизма [4 + 2] циклоприсоединение из изопрен с малеиновый ангидрид.[38] Предыдущие исследования Гаевского на изотопически обогащенных материалах наблюдали результаты КИЭ, которые предполагали асинхронное переходное состояние, но всегда были согласованными, в пределах ошибки, для идеально синхронного механизм реакции.[43]

Эта работа Синглтона и др. установил измерение нескольких 13
C
KIE в рамках единственного эксперимента. Эти 2
ЧАС
и 13
C
Измерения KIE, определенные при естественном изобилии, показали, что «внутренние» водороды диена испытывают более выраженную 2
ЧАС
KIE, чем «внешние» водороды », и C1 и C4 испытывают значительный KIE. Эти ключевые наблюдения предполагают асинхронный механизм реакции для циклоприсоединение из изопрен с малеиновый ангидрид.
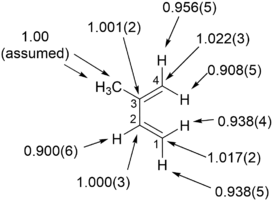
Ограничения для определения KIE в естественном изобилии с использованием ЯМР заключаются в том, что извлеченный материал должен иметь подходящее количество и чистоту для анализа ЯМР (интересующий сигнал должен отличаться от других сигналов), интересующая реакция должна быть необратимой и механизм реакции не должно изменяться в течение химическая реакция.
Экспериментальные детали для использования количественного одноимпульсного ЯМР для измерения кинетического изотопного эффекта при естественном содержании следующим образом: эксперимент необходимо проводить в количественных условиях, включая время релаксации 5 Тл.1, измеренный угол поворота 90 °, цифровое разрешение не менее 5 точек по пику и сигнал: шум больше 250. Необработанный FID заполняется нулями по крайней мере до 256 К точек перед преобразованием Фурье. Спектры ЯМР фазированы и затем обрабатываются с поправкой нулевого порядка базовой линии без какой-либо поправки на наклон. Интегрирование сигналов определяется численно с минимальным допуском для каждого интегрированного сигнала.[38][требуется разъяснение ]
Примеры выяснения механизма металлоорганических реакций
Коллетто и другие. разработал региоселективный -арилирование бензо [b] тиофенов при комнатной температуре с арилиодидами в качестве партнеров по связыванию и стремились понять механизм этой реакции, выполнив измерения кинетического изотопного эффекта естественного содержания с помощью одноимпульсного ЯМР.[44]




Наблюдение за первичным 13Изотопный эффект C на C3, обратный 2H изотопный эффект, вторичный 13Изотопный эффект C на C2 и отсутствие 2Изотопный эффект H в отведении C2 Colletto и другие. предложить механизм реакции типа Хека для региоселективного -арилирование бензо [b] тиофенов при комнатной температуре с арилиодидами в качестве партнеров связывания.[44]
Мороз et al. стремился понять влияние Кислота Льюиса добавок по механизму энантиоселективной активации связи C-N, катализируемой палладием, с использованием измерений кинетического изотопного эффекта естественной распространенности с помощью одноимпульсного ЯМР.[45]


Главная 13Кинетический изотопный эффект C, наблюдаемый в отсутствие BPh3 предполагает механизм реакции с ограничением скорости цис-окисления в связь C – CN цианоформамид. Добавление BPh3 вызывает относительное уменьшение наблюдаемого 13C кинетический изотопный эффект, который привел Фроста et al. предложить изменение стадии ограничения скорости с цис-окисления на координацию палладия с цианоформамидом.[45]
DEPT-55 ЯМР
Хотя измерения кинетического изотопного эффекта при естественном изобилии являются мощным инструментом для понимания механизмов реакции, количество материала, необходимое для анализа, может сделать этот метод недоступным для реакций, в которых используются дорогие реагенты или нестабильные исходные материалы. Чтобы смягчить эти ограничения, Якобсен с коллегами разработали 1H к 13Перенос поляризации углерода как средство сокращения времени и материалов, необходимых для измерений кинетического изотопного эффекта при естественном изобилии. В улучшение без искажений за счет передачи поляризации (DEPT) использует преимущества большего гиромагнитное отношение из 1H более 13C для теоретического повышения чувствительности измерения в 4 раза или уменьшения времени эксперимента в 16 раз. Этот метод измерения кинетических изотопов естественного содержания подходит для анализа реакций, содержащих нестабильные исходные материалы, а также катализаторы или продукты, которые относительно дороги.[46]
Якобсен с соавторами определили гликозилирование галактозы, катализируемое тиомочевиной, как реакцию, которая удовлетворяет обоим вышеупомянутым критериям (дорогие материалы и нестабильные субстраты), и является реакцией с плохо изученным механизмом.[47] Гликозилирование - это частный случай нуклеофильного замещения, которому не хватает четкого определения между SN1 и SN2 механистический персонаж. Присутствие кислорода рядом с местом смещения (например, C1) может стабилизировать положительный заряд. Эта стабилизация заряда может привести к тому, что любой потенциальный согласованный путь станет асинхронным и приближается к промежуточным продуктам с оксокарбениевым характером SN1 механизм гликозилирования.


Якобсен и его коллеги наблюдали небольшие нормальные КИЭ в С1, С2 и С5, что указывает на значительный характер оксокарбения в переходном состоянии и асинхронный механизм реакции с большой степенью разделения зарядов.
Масс-спектрометрия изотопного отношения
Высокая точность масс-спектрометрия изотопного отношения (IRMS) - еще один метод измерения кинетическое фракционирование из изотопы для измерений KIE естественного обилия. Видлански и его коллеги продемонстрировали 34
S
KIE при измерениях естественного обилия для гидролиз из сульфат моноэфиры. Их наблюдение за большим KIE предполагает, что разрыв облигаций S-O контролирует процентную ставку и, вероятно, исключает партнерство. механизм реакции.[48]

Основным ограничением для определения KIE в естественном изобилии с помощью IRMS является требуемая сайт-селективная деградация без изотопного фракционирования до анализируемой небольшой молекулы, что является нетривиальной задачей.[38]
Тематические исследования
Эффекты первичных изотопов водорода
Первичные кинетические изотопные эффекты водорода относятся к случаям, в которых связь с меченным изотопом водородом образуется или разрывается на стадии реакции, определяющей скорость и / или продукт.[5] Это наиболее часто измеряемые кинетические изотопные эффекты, и большая часть ранее рассмотренной теории относится к первичным кинетическим изотопным эффектам. Когда есть достаточные доказательства того, что перенос меченого водорода происходит на определяющей скорость стадии реакции, если достаточно большой наблюдается кинетический изотопный эффект, например kH / kD, по крайней мере, 5-6 или kH / kT около 10-13 при комнатной температуре, вполне вероятно, что перенос водорода является линейным и что водород довольно симметрично расположен в переходном состоянии. Как правило, невозможно комментировать туннельные вклады в наблюдаемый изотопный эффект, если эффект не очень велик. Если первичный кинетический изотопный эффект не так велик, он обычно считается показателем значительного вклада движения тяжелого атома в координату реакции, хотя это также может означать, что перенос водорода происходит по нелинейному пути.[8]
Вторичные изотопные эффекты водорода
Вторичный изотопный эффект водорода или вторичный кинетический изотопный эффект (SKIE) возникает в случаях, когда изотопное замещение удалено от разрываемой связи. Тем не менее удаленный атом влияет на внутренние колебания системы, которые через изменение нулевой энергии (ZPE) влияют на скорость химических реакций.[49] Такие эффекты выражаются в виде отношения скорости легкого изотопа к скорости тяжелого изотопа и могут быть «нормальными» (отношение больше или равно 1) или «обратными» (отношение меньше 1) эффектами.[50] SKIE определяются как α, β (и т. д.) вторичные изотопные эффекты, где такие префиксы относятся к положению изотопного замещения относительно реакционного центра (см. альфа и бета углерод ).[51] Префикс α относится к изотопу, связанному с реакционным центром, а префикс β относится к изотопу, связанному с атомом, соседним с реакционным центром, и так далее.
В физической органической химии SKIE обсуждается с точки зрения электронные эффекты такие как индукция, гибридизация связей или сверхсопряжение.[52] Эти свойства определяются распределением электронов и зависят от колебательно-усредненной длины связи и углов, на которые изотопное замещение не сильно влияет. Таким образом, использование термина «электронный изотопный эффект», хотя оно и является законным, не рекомендуется, поскольку его можно неверно истолковать, предполагая, что изотопный эффект имеет электронную природу, а не колебательный.[51]
SKIE можно объяснить изменениями орбитальной гибридизации. Когда гибридизация атома углерода изменяется с sp3 к sp2влияет на ряд колебательных мод (растяжения, изгиб в плоскости и вне плоскости). Изгиб в плоскости и вне плоскости в пр3 гибридизованный углерод похожи по частоте из-за симметрии sp3 гибридизированный углерод. В зр2 из гибридизированного углерода изгиб в плоскости намного жестче, чем изгиб вне плоскости, что приводит к большой разнице в частоте, ZPE и, таким образом, SKIE (который существует, когда существует разница в ZPE реагента и переходного состояния).[20]Теоретическое максимальное изменение, вызванное разностью частот изгиба, было рассчитано как 1,4.[20]
Когда углерод претерпевает реакцию, изменяющую его гибридизацию с sp3 к sp2, константа изгибающей силы вне плоскости в переходном состоянии тем меньше, чем больше sp2 характер и "нормальный" SKIE наблюдается с типичными значениями от 1,1 до 1,2.[20] И наоборот, когда гибридизация углерода изменяется от sp2 к sp3, константы силы изгиба вне плоскости в переходном состоянии увеличиваются, и наблюдается обратный SKIE с типичными значениями от 0,8 до 0,9.[20]
В более общем смысле SKIE для обратимых реакций может быть "нормальным" в одном направлении и "обратным" в другом, если связывание в переходном состоянии находится на полпути по жесткости между подложкой и продуктом, или они могут быть "нормальными" в обоих направлениях, если связывание слабее в переходное состояние или "обратное" в обоих направлениях, если связь в переходном состоянии сильнее, чем в любом из реагентов.[50]
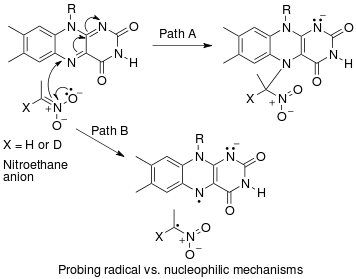
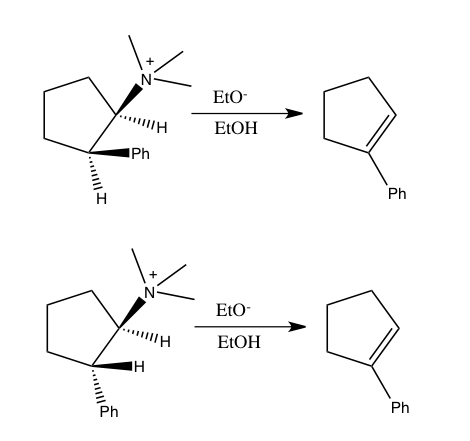
Пример «обратного» вторичного кинетического изотопного эффекта α можно увидеть в работе Фитцпатрика и Курца, которые использовали такой эффект, чтобы различить два предложенных пути реакции оксидаза d-аминокислот с нитроалкан анионы.[53] Путь A включает нуклеофильную атаку на кофермент FAD, в то время как путь B включает промежуточное соединение свободных радикалов. Поскольку путь A приводит к гибридизации с промежуточным изменением углерода из sp2 к sp3 ожидается "обратная" SKIE. Если происходит путь B, то SKIE не должно наблюдаться, поскольку промежуточный продукт свободных радикалов не изменяет гибридизацию. Наблюдалось значение SKIE 0,84, и путь A подтверждался, как показано на схеме ниже.
Другой пример SKIE - окисление бензиловых спиртов диметилдиоксираном, где были предложены три переходных состояния для различных механизмов. Опять же, рассмотрев, как и если бы атомы водорода были вовлечены в каждый из них, исследователи предсказали, ожидают ли они эффекта изотопного замещения их. Затем анализ экспериментальных данных для реакции позволил им выбрать, какой путь наиболее вероятно основан на наблюдаемом изотопном эффекте.[54]
Эффекты вторичных изотопов водорода от метиленовых водородов также использовались, чтобы показать, что перегруппировка Коупа в 1,5-гексадиене идет по согласованному пути перегруппировки связей, а не по одному из альтернативно предлагаемых аллильных радикалов или 1,4-диильных путей, которые все являются представлен на следующей схеме.[55]

Альтернативные механизмы для перегруппировки Коупа 1,5-гексадиена: (сверху вниз), аллильный радикал, синхронные согласованные пути и пути 1,4-диила. Обнаружено, что преобладающим путем является средний путь, который имеет шесть делокализованных π-электронов, соответствующих ароматическому промежуточному соединению.[55]
Стерические изотопные эффекты
 |

Стерический изотопный эффект - это SKIE, который не включает разрыв или образование связей. Этот эффект объясняется разной амплитудой колебаний изотопологи.[56] Примером такого эффекта является рацемизация 9,10-дигидро-4,5-диметилфенантрена.[57] Меньшая амплитуда колебаний для дейтерия по сравнению с водородом в связях C – H (углерод – водород), C – D (углерод – дейтерий) приводит к меньшему ван-дер-ваальсовому радиусу или эффективному размеру в дополнение к разнице в ZPE между два. Когда существует большая эффективная масса молекул, содержащих одну над другой, это может проявляться стерическим влиянием на константу скорости. В приведенном выше примере дейтерий рацемизируется быстрее, чем изотополог водорода, что приводит к стерическому изотопному эффекту. Модель стерического изотопного эффекта была разработана Бартеллом.[58] Эффект стерического изотопа обычно невелик, если преобразования не проходят через переходное состояние с серьезными стерическими препятствиями, как в процессе рацемизации, показанном выше.
Другой пример стерического изотопного эффекта - это реакция удаления скольжения ротаксанов. Изотоп дейтерия, благодаря его меньшему эффективному размеру, позволяет более легкому прохождению стопоров через макроцикл, что приводит к более высокой скорости удаления дейтерированного ротаксаны.[59]
Обратные кинетические изотопные эффекты
Известны реакции, когда реагируют дейтерированные частицы. Быстрее чем недейтерированный аналог, и говорят, что в этих случаях проявляются обратные кинетические изотопные эффекты (IKIE). IKIE часто наблюдаются в восстановительное устранение алкилгидридов металлов, например (Мне2NCH2CH2NMe2 ) PtMe (H). В таких случаях связь C-D в переходном состоянии агостик разновидностей, высоко стабилизирован относительно связи C – H.[нужна цитата ]
Обратный эффект также может иметь место в многоступенчатой реакции, если общая константа скорости зависит от предварительное равновесие до этап определения ставки который имеет обратный равновесный изотопный эффект. Например, ставки кислотно-катализированный реакции обычно в 2-3 раза больше для реакций в D2O катализируется D3О+ чем для аналогичных реакций в H2O катализируется H3О+[4]:433 Это можно объяснить механизмом специфический водородно-ионный катализ реагента R на H3О+ (или D3О+).
- ЧАС3О+ + R ⇌ RH+ + H2О
- RH+ + H2O → H3О+ + P
Тогда скорость образования продуктов равна d [P] / dt = k2[RH+] = k2K1[ЧАС3О+] [R] = kНаблюдения[ЧАС3О+][Р]. На первом этапе H3О+ обычно более сильная кислота, чем RH+. Дейтерирование сдвигает равновесие в сторону более прочно связанных кислотных форм RD+ в котором влияние дейтерирования на нулевую колебательную энергию больше, так что дейтерированная константа равновесия K1D больше K1H. Этот равновесный изотопный эффект на первом этапе обычно перевешивает кинетический изотопный эффект на втором этапе, так что существует очевидный обратный изотопный эффект и наблюдаемая общая константа скорости kНаблюдения = k2K1 уменьшается.[4]:433
Кинетические изотопные эффекты водорода в растворителе
Чтобы изотопные эффекты растворителя можно было измерить, конечная часть растворителя должна иметь изотопный состав, отличный от остального. Следовательно, должны быть доступны большие количества менее распространенных изотопных соединений, ограничивая наблюдаемые изотопные эффекты растворителя изотопными замещениями с участием водорода. Обнаруживаемые кинетические изотопные эффекты возникают только тогда, когда растворенные вещества обмениваются водородом с растворителем или когда существует определенное взаимодействие растворенного вещества и растворителя рядом с местом реакции. Оба эти явления являются общими для протонных растворителей, в которых водород является обменным, и они могут образовывать диполь-дипольные взаимодействия или водородные связи с полярными молекулами.[8]
Изотопные эффекты углерода-13
Большинство органических реакций включают разрыв и образование связей с углеродом; таким образом, можно ожидать обнаруживаемых изотопных эффектов углерода. Когда 13C используется в качестве метки, однако изменение массы изотопа составляет всего ~ 8%, что ограничивает наблюдаемые кинетические изотопные эффекты гораздо меньшими значениями, чем те, которые наблюдаются при изотопных эффектах водорода.
Компенсация вариаций в 13C естественное изобилие
Часто самый большой источник ошибок в исследовании, который зависит от естественного содержания углерода, - это незначительное изменение естественного содержания углерода. 13C самим изобилием. Такие вариации возникают из-за того, что исходные материалы, используемые в реакции, сами являются продуктами некоторых других реакций, которые обладают кинетическими изотопными эффектами и соответствующими изотопными обогащениями в продуктах. Чтобы компенсировать эту ошибку при использовании ЯМР-спектроскопии для определения кинетического изотопного эффекта, были предложены следующие рекомендации:[39][40]
- Выберите углерод, удаленный от реакционного центра, который будет служить эталоном, и предположите, что он не оказывает кинетического изотопного эффекта в реакции.
- В исходном материале, который не подвергался какой-либо реакции, определяют отношения интегралов пиков других углеродных ЯМР к таковому у эталонного углерода.
- Получите такие же соотношения для углерода в образце исходного материала после того, как он подвергся некоторой реакции.
- Отношение последних отношений к первым отношениям дает R / R0.
Если соблюдать эти, а также некоторые другие меры предосторожности, перечисленные Янковским, можно достичь кинетических изотопных эффектов с точностью до трех знаков после запятой.[39][40]
Изотопные эффекты с элементами тяжелее углерода
Интерпретация изотопных эффектов углерода обычно осложняется одновременным образованием и разрывом связей с углеродом. Даже реакции, которые включают только разрыв связи с углеродом, такие как SN1, включают усиление оставшихся связей с углеродом. Во многих таких реакциях изотопные эффекты уходящей группы, как правило, легче интерпретировать. Например, реакции замещения и элиминирования, в которых хлор действует как уходящая группа, удобно интерпретировать, особенно потому, что хлор действует как одноатомная разновидность без внутренней связи, усложняющей координату реакции, и имеет два стабильных изотопа, 35Cl и 37Cl, оба с высоким содержанием. Основной проблемой при интерпретации таких изотопных аффектов является сольватация уходящей группы.[8]
Из-за экспериментальных неопределенностей измерение изотопного эффекта может повлечь за собой значительную неопределенность. Часто изотопные эффекты определяются путем дополнительных исследований ряда изотопомеров. Соответственно, весьма полезно сочетать изотопные эффекты водорода с изотопными эффектами тяжелых атомов. Например, определение изотопного эффекта азота наряду с изотопным эффектом водорода было использовано, чтобы показать, что реакция иона 2-фенилэтилтриметиламмония с этоксидом в этаноле при 40 ° C следует по механизму E2, в отличие от альтернативных несогласованных механизмов. Этот вывод был сделан после того, как было показано, что эта реакция дает изотопный эффект азота, k14/k151,0133 ± 0,0002 вместе с кинетическим изотопным эффектом водорода 3,2 на выходящем водороде.[8]
Точно так же сочетание изотопных эффектов азота и водорода было использовано, чтобы показать, что син-элиминирование простых солей аммония также следует согласованному механизму, который ранее обсуждался. В следующих двух реакциях иона 2-фенилциклопентилтриметиламмония с этоксидом, обе из которых дают 1-фенилциклопентен, оба изомера проявляют изотопный эффект азота. k14/k15 при 60 ° С. Хотя реакция транс-изомера, которая следует за син-элиминированием, имеет меньший кинетический изотопный эффект азота (1,0064) по сравнению с цис-изомером, который подвергается антиэлиминированию (1,0108), оба результата достаточно велики, чтобы указывать на ослабление связи CN. в переходном состоянии, которое произойдет в согласованном процессе.
Другие примеры
Поскольку кинетические изотопные эффекты возникают из-за различий в массах изотопов, наибольшие наблюдаемые кинетические изотопные эффекты связаны с изотопными замещениями водорода дейтерием (увеличение массы на 100%) или тритием (увеличение массы на 200%). Кинетические изотопные эффекты от отношения изотопных масс могут достигать 36,4 при использовании мюонов. Они произвели легчайший атом водорода, 0.11H (0,113 а.е.м.), в котором электрон вращается вокруг положительного мюона (μ+) «ядро», имеющее массу 206 электронов. Они также приготовили аналог самого тяжелого атома водорода, заменив один электрон в гелии отрицательным мюоном (μ−) с образованием Heμ с атомной массой 4,116 а.е.м. Поскольку отрицательный мюон намного тяжелее электрона, он вращается гораздо ближе к ядру, эффективно экранируя один протон, заставляя Heμ вести себя как 4.1H. С этими экзотическими видами реакция H с 1ЧАС2 был исследован. Константы скорости реакции самых легких и тяжелых аналогов водорода с 1ЧАС2 затем использовались для расчета k0.11/k4.1 кинетический изотопный эффект, при котором разница изотопных масс в 36,4 раза. В этой реакции изотопное замещение приводит к обратному кинетическому изотопному эффекту, и авторы сообщают о кинетическом изотопном эффекте всего 1,74 × 10−4, что является наименьшим из когда-либо зарегистрированных кинетических изотопных эффектов.[60]
Кинетический изотопный эффект приводит к определенному распределению изотопов дейтерия в природных продуктах, в зависимости от того, каким путем они были синтезированы в природе. Поэтому с помощью ЯМР-спектроскопии легко определить, был ли спирт в вине сброжен из глюкоза, или из незаконно добавленных сахароза.
Другой механизмы реакции что было выяснено с помощью кинетического изотопного эффекта, является галогенирование из толуол:[61]
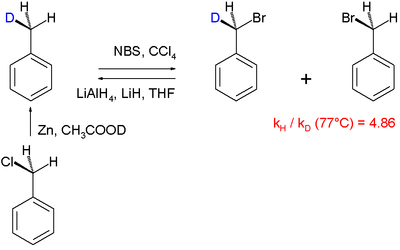
В этом конкретном исследовании "внутримолекулярного KIE" бензиловый водород подвергается радикальное замещение бромом с использованием N-бромосукцинимид как бромирующий агент. Было обнаружено, что PhCH3 броматы в 4,86 раза быстрее, чем PhCD3. Большой KIE 5,56 связан с реакцией кетоны с бром и едкий натр.[62]

В этой реакции лимитирующей стадией является образование энолировать депротонированием кетона. В этом исследовании KIE рассчитывается из константы скорости реакции для обычного 2,4-диметил-3-пентанона и его дейтерированного изомера по оптическая плотность измерения.
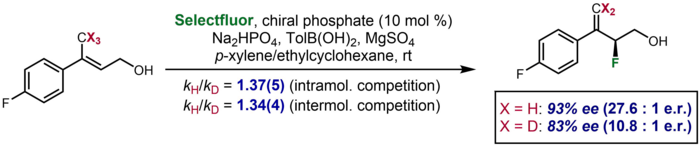
При асимметричном катализе в редких случаях кинетический изотопный эффект проявляется как существенное различие в энантиоселективности, наблюдаемой для дейтерированного субстрата по сравнению с недейтерированным. Один пример был описан Toste и соавторами, в котором дейтерированный субстрат давал энантиоселективность 83% ee по сравнению с 93% ee для недейтерированного субстрата. Эффект был предпринят для подтверждения дополнительных данных KIE о меж- и внутримолекулярной конкуренции, которые предполагали разрыв связи C-H / D на стадии энантиодетерминации.[63]
Смотрите также
- Кроссовер эксперимент (химия)
- Константа равновесия # Эффект изотопного замещения
- Магнитный изотопный эффект
- Механизм реакции
- Переходное кинетическое фракционирование изотопов
Рекомендации
- ^ а б c d е Westaway KC (2006). «Использование кинетических изотопических эффектов для определения структуры переходных состояний SN2 реакции ». Успехи физико-органической химии. 41: 217–273. Дои:10.1016 / S0065-3160 (06) 41004-2.
- ^ Линн К.Р., Янквич ЧП (5 августа 1961 г.). «Изотопное фракционирование на метилуглероде в реакциях иона цианида с метилхлоридом и метилбромидом». Журнал Американского химического общества. 83 (15): 3220–3223. Дои:10.1021 / ja01476a012.
- ^ а б c d е ж Аткинс П., де Паула Дж (2006). Физическая химия Аткинса (8-е изд.). Oxford University Press. стр.286 –288, 816–818. ISBN 978-0-19-870072-2.
- ^ а б c d е ж грамм час Лайдлер К.Дж. (1987). Химическая кинетика (3-е изд.). Харпер и Роу. ISBN 978-0-06-043862-3.
- ^ а б c d Симмонс Э.М., Хартвиг Дж. Ф. (март 2012 г.). «Об интерпретации кинетических изотопных эффектов дейтерия в функционализации связи C – H комплексами переходных металлов». Angewandte Chemie International Edition. 51 (1): 3066–72. Дои:10.1002 / anie.201107334. PMID 22392731.
- ^ Poirier RA, Wang Y, Westaway KC (март 1994). "Теоретическое исследование связи между кинетическими изотопными эффектами вторичного альфа-дейтерия и структурой серы.N2 Переходные состояния ». Журнал Американского химического общества. 116 (6): 2526–2533. Дои:10.1021 / ja00085a037.
- ^ а б c d е Бунсел Э, Ли CC (1977). Изотопы в катионных реакциях. Изотопы в органической химии. 5. Амстердам: Эльзевир. ISBN 978-0-444-41927-9. OCLC 867217247.
- ^ а б c d е ж грамм час я Меландер Л., Сондерс WH (1980). Скорости реакций изотопных молекул. Нью-Йорк: Вили.
- ^ Bigeleisen J, Wolfsberg M (январь 1957 г.). «Теоретические и экспериментальные аспекты изотопных эффектов в химической кинетике». Успехи химической физики. 1: 15–76.
- ^ Если мюоний (μ+е–) рассматривается как изотоп водорода, то в принципе возможны даже более крупные КИЭ. Однако исследования с участием мюония ограничены коротким периодом полураспада мюона (22 микросекунды) (см. Villà J, Corchado JC, González-Lafont A, Lluch JM, Truhlar DG (ноябрь 1998 г.). «Объяснение кинетических изотопных эффектов дейтерия и мюония при присоединении атома водорода к олефину». Журнал Американского химического общества. 120 (46): 12141–2. Дои:10.1021 / ja982616i. для примера kМу/kЧАС изотопный эффект.)
- ^ Это соглашение существует как для удобства номенклатуры, так и для отражения того, как кинетические изотопные эффекты дейтерия обычно изучаются экспериментально: хотя дейтерий имеет санкционированный ИЮПАК символ D = 2H не существует общего символа, конкретно относящегося к протию (1ЧАС). Тем не менее, оказалось полезным иметь метки для обозначения соответствующих констант скорости изотопологов, содержащих протий или дейтерий, поэтому kЧАС и kD, соответственно, обычно использовались. Более того, величина кинетического изотопного эффекта может быть выражена как kЧАС/kD. Это обозначение согласуется с тем фактом, что экспериментально кинетические изотопные эффекты дейтерия измеряются путем сравнения скорости реакции обогащенного дейтерием исходного материала со скоростью реакции необогащенного исходного материала, содержащего водород в естественном количестве. Это почти всегда верно, поскольку протий составляет 99,9885% природного водорода, поэтому обычно нет необходимости в дальнейшем обеднении дейтерия в исходном материале для получения «обогащенного протием» образца. В совокупности обозначения и экспериментальная установка привели к общей концепции дейтерия как «заместителя», который заменяет «обычный» водород в исследовании изотопного эффекта.
- ^ Bigeleisen J (август 1949 г.). «Относительные скорости реакций изотопных молекул». Журнал химической физики. 17 (8): 675–678. Bibcode:1949ЖЧФ..17..675Б. Дои:10.1063/1.1747368.
- ^ а б c Лоури TH, Ричардсон KS (1987). Механизм и теория в органической химии (3-е изд.). Нью-Йорк: Харпер и Роу. стр.256. ISBN 978-0-06-044084-8. OCLC 14214254.
- ^ Карпентер Б.К. (1984). Определение механизмов органических реакций. Нью-Йорк: Вили. п. 86. ISBN 978-0-471-89369-1. OCLC 9894996.
- ^ Карпентер Б.К. (февраль 2010 г.). «Кинетические изотопные эффекты: открытие нетрадиционного». Химия природы. 2 (2): 80–2. Bibcode:2010НатЧ ... 2 ... 80С. Дои:10.1038 / nchem.531. PMID 21124393.
- ^ Кэрролл FA (2010). Перспективы структуры и механизма в органической химии (2-е изд.). Хобокен, штат Нью-Джерси: Джон Вили. ISBN 978-0-470-27610-5. OCLC 286483846.
- ^ Кварт Х (1 декабря 1982 г.). «Температурная зависимость первичного кинетического изотопного эффекта водорода как механистический критерий». Отчеты о химических исследованиях. 15 (12): 401–408. Дои:10.1021 / ar00084a004. ISSN 0001-4842.
- ^ Streitwieser A, Jagow RH, Fahey RC, Suzuki S (май 1958 г.). «Кинетические изотопные эффекты в ацетолизе дейтерированных циклопентилтозилатов1, 2». Журнал Американского химического общества. 80 (9): 2326–32. Дои:10.1021 / ja01542a075.
- ^ Суэйн К.Г., Стиверс Е.К., Ройвер-младший Дж. Ф., Шаад Л.Дж. (1 ноября 1958 г.). «Использование эффектов изотопа водорода для идентификации атакующего нуклеофила при енолизации кетонов, катализируемых уксусной кислотой». Журнал Американского химического общества. 80 (21): 5885–5893. Дои:10.1021 / ja01554a077.
- ^ а б c d е ж грамм час я j Анслин Э.В., Догерти Д.А. (2006). Современная физико-органическая химия. Книги университетских наук. стр.428 –437. ISBN 978-1-891389-31-3.
- ^ Разауи М (2003). Квантовая теория туннелирования. Всемирный научный. ISBN 978-981-238-019-7.
- ^ Силби Р.Дж., Олберти Р.А., Бавенди М.Г. (2005). Физическая химия. Джон Уайли и сыновья. С. 326–338. ISBN 978-0-471-21504-2.
- ^ Боргис Д., Хайнс Дж. Т. (1993). «Динамическая теория скоростей туннельного переноса протона в растворе: общая формулировка». Химическая физика. 170 (3): 315–346. Bibcode:1993CP .... 170..315B. Дои:10.1016 / 0301-0104 (93) 85117-Q.
- ^ а б Кришталик Л.И. (май 2000 г.). «Механизм переноса протона: очерк». Biochimica et Biophysica Acta. 1458 (1): 6–27. Дои:10.1016 / S0005-2728 (00) 00057-8. PMID 10812022.
- ^ Зуев П.С., Шеридан Р.С., Альбу Т.В., Трухлар Д.Г., Хроват Д.А., Борден В.Т. (февраль 2003 г.). «Углеродное туннелирование из одиночного квантового состояния». Наука. 299 (5608): 867–70. Bibcode:2003Наука ... 299..867Z. Дои:10.1126 / science.1079294. PMID 12574623.
- ^ Fujisaki N, Ruf A, Gaeumann T (1987). «Туннельные эффекты в реакциях переноса атома водорода, как изучено с помощью температурной зависимости кинетических изотопных эффектов дейтерия водорода». Журнал физической химии. 91 (6): 1602–1606. Дои:10.1021 / j100290a062.
- ^ Льюис ES, Фундерберк Л. (1967). «Скорости и изотопные эффекты в переходах протонов от 2-нитропропана к пиридиновым основаниям». Журнал Американского химического общества. 89 (10): 2322–2327. Дои:10.1021 / ja00986a013.
- ^ Дьюар MJ, Хили EF, Руис JM (1988). «Механизм 1,5-сигматропного водородного сдвига в 1,3-пентадиене». Журнал Американского химического общества. 110 (8): 2666–2667. Дои:10.1021 / ja00216a060.
- ^ фон Деринг В., Чжао Х (июль 2006 г.). «Влияние дейтерия на кинетику сдвига 1,5-водорода заблокированного цизоидом 1,3 (Z) -пентадиена, 2-метил-10-метиленбицикло [4.4.0] дец-1-ена: доказательства туннелирования? ". Журнал Американского химического общества. 128 (28): 9080–5. Дои:10.1021 / ja057377v. PMID 16834382.
- ^ В этом исследовании KIE измеряется чувствительными протонный ЯМР. Экстраполированный KIE при 25 ° C составляет 16,6, но предел погрешности высок.
- ^ Коэн А., Клинман Дж. П. (июль 1999 г.). «Водородное туннелирование в биологии». Химия и биология. 6 (7): R191-8. Дои:10.1016 / S1074-5521 (99) 80058-1. PMID 10381408.
- ^ Wilde TC, Blotny G, Pollack RM (май 2008 г.). «Экспериментальные доказательства связанного движения с ферментами / квантово-механического туннелирования водорода с помощью кетостероидизомеразы». Журнал Американского химического общества. 130 (20): 6577–85. Дои:10.1021 / ja0732330. PMID 18426205.
- ^ Трулар Д.Г., Гао Дж., Альгамбра К., Гарсия-Вилока М., Корчадо Дж., Санчес М., Вилла Дж. (2002). «Включение квантовых эффектов в моделирование кинетики ферментов». Отчеты о химических исследованиях. 35 (6): 341–349. Дои:10.1021 / ar0100226.
- ^ Коэн, А; Клинман, Дж. П (1998). «Ферментный катализ: за пределами классических парадигм». Отчеты о химических исследованиях. 31 (7): 397–404. Дои:10.1021 / ar9701225.
- ^ Магги Ф, Райли У. Дж. (2010). «Математическая обработка видообразования и фракционирования изотопологов и изотопомеров в биохимической кинетике». Geochimica et Cosmochimica Acta. 74 (6): 1823. Bibcode:2010GeCoA..74.1823M. Дои:10.1016 / j.gca.2009.12.021.
- ^ Ньюолл, А. Раймонд; Хейс, Джон; Бетелл, Дональд (1 января 1974 г.). "Промежуточные продукты разложения алифатических диазосоединений. Часть XI. Механические исследования реакции дифенилметилена с аминами в растворе". Журнал химического общества, Perkin Transactions 2. 0 (11): 1307–1312. Дои:10.1039 / P29740001307. ISSN 1364-5471.
- ^ Бунсел Э, Ли CC (1977). Углерод-13 в органической химии. Изотопы в органической химии. 3. Амстердам: Эльзевир. ISBN 978-0-444-41472-4. OCLC 606113159.
- ^ а б c d Синглтон Д.А., Томас А.А. (сентябрь 1995 г.). «Высокоточное одновременное определение множественных малых кинетических изотопных эффектов при естественном изобилии». Журнал Американского химического общества. 117 (36): 9357–9358. Дои:10.1021 / ja00141a030.
- ^ а б c Янковский С. (январь 2009 г.). «Применение ЯМР-спектроскопии в исследованиях изотопных эффектов». Годовые отчеты по ЯМР-спектроскопии. 68: 149–191. Дои:10.1016 / S0066-4103 (09) 06803-3. ISBN 9780123810410.
- ^ а б c Кван Э. «Заметки к курсу CHEM 106 - Лекция 14 - Вычислительная химия» (PDF). Получено 2 ноября 2013.
- ^ Мартин GJ, Мартин ML (1984). «Мечение дейтерием на естественном уровне изобилия, как изучено с помощью высокопольного количественного 2H ЯМР». Буквы Тетраэдра. 22 (36): 3525–3528. Дои:10.1016 / s0040-4039 (01) 81948-1.
- ^ а б Паскаль мл. Р. А., Баум М. В., Вагнер К. К., Роджерс Л. Р. (сентябрь 1984 г.). «Измерение кинетических изотопных эффектов дейтерия в органических реакциях методом ЯМР-спектроскопии естественного содержания дейтерия». Журнал Американского химического общества. 106 (18): 5377–5378. Дои:10.1021 / ja00330a071.
- ^ Гаевский Дж. Дж., Петерсон КБ, Кагель Дж. Р., Хуанг Ю. Дж. (Декабрь 1989 г.). «Изменение структуры переходного состояния в реакции Дильса-Альдера из-за вторичных кинетических изотопных эффектов дейтерия. Реакция почти симметричных диенов и диенофилов почти синхронна». Журнал Американского химического общества. 111 (25): 9078–9081. Дои:10.1021 / ja00207a013.
- ^ а б Коллетто С., Ислам С., Хулиа-Эрнандес Ф., Ларроса I (февраль 2016 г.). «Прямое β-арилирование тиофенов и бензо [b] тиофенов при комнатной температуре и кинетические доказательства пути типа Хека». Журнал Американского химического общества. 138 (5): 1677–83. Дои:10.1021 / jacs.5b12242. ЧВК 4774971. PMID 26788885.
- ^ а б Frost GB, Serratore NA, Ogilvie JM, Douglas CJ (апрель 2017 г.). "Механистическая модель энантиоселективного внутримолекулярного цианоамидирования алкенов посредством активации связи C-CN, катализируемой палладием". Журнал органической химии. 82 (7): 3721–3726. Дои:10.1021 / acs.joc.7b00196. ЧВК 5535300. PMID 28294618.
- ^ Кван Э., Парк Й., Бессер Х.А., Андерсон Т.Л., Якобсен Э.Н. (январь 2017 г.) «Измерения кинетического изотопного эффекта 13C с помощью передачи поляризации». Журнал Американского химического общества. 139 (1): 43–46. Дои:10.1021 / jacs.6b10621. ЧВК 5674980. PMID 28005341.
- ^ Park Y, Харпер KC, Kuhl N, Kwan EE, Liu RY, Jacobsen EN (январь 2017 г.). «Макроциклические бис-тиомочевины катализируют реакции стереоспецифического гликозилирования». Наука. 355 (6321): 162–166. Bibcode:2017Научный ... 355..162P. Дои:10.1126 / science.aal1875. ЧВК 5671764. PMID 28082586.
- ^ Берлингем Б.Т., Пратт Л.М., Дэвидсон Э.Р., Шайнер В.Дж., Фонг Дж., Видлански Т.С. (октябрь 2003 г.). «Эффект изотопа 34S на гидролиз сульфатного эфира: механистические последствия». Журнал Американского химического общества. 125 (43): 13036–7. Дои:10.1021 / ja0279747. PMID 14570471.
- ^ Хенниг С., Освальд Р. Б., Шматц С. (март 2006 г.). «Вторичный кинетический изотопный эффект при нуклеофильном замещении: квантово-механический подход». Журнал физической химии A. 110 (9): 3071–9. Bibcode:2006JPCA..110.3071H. Дои:10.1021 / jp0540151. PMID 16509628.
- ^ а б Cleland WW (декабрь 2003 г.). «Использование изотопных эффектов для определения ферментативных механизмов». Журнал биологической химии. 278 (52): 51975–84. Дои:10.1074 / jbc.X300005200. PMID 14583616.
- ^ а б ИЮПАК, Сборник химической терминологии, 2-е изд. («Золотая книга») (1997). Исправленная онлайн-версия: (2006–) "Вторичный изотопный эффект ". Дои:10.1351 / goldbook.S05523
- ^ «Определение изотопного эффекта, вторичного». Химический словарь. Chemitool.
- ^ Курц К.А., Фицпатрик П.Ф. (1997). «Влияние pH и вторичных кинетических изотопов на реакцию D-аминокислотной оксидазы с нитроалкановыми анионами: доказательства прямого воздействия карбанионов на флавин». Журнал Американского химического общества. 119 (5): 1155–1156. Дои:10.1021 / ja962783n.
- ^ Ангелис Ю.С., Хатзакис Н.С., Смоноу I, Орфанопулос М. (2006). «Окисление бензиловых спиртов диметилдиоксираном. Вопрос о согласованных и ступенчатых механизмах, исследуемых кинетическими изотопными эффектами». Буквы Тетраэдра. 42 (22): 3753–3756. Дои:10.1016 / S0040-4039 (01) 00539-1.
- ^ а б Houk KN, Gustafson SM, Black KA (октябрь 1992 г.). «Теоретические вторичные кинетические изотопные эффекты и интерпретация геометрии переходного состояния. 1. Перегруппировка Коупа». Журнал Американского химического общества. 114 (22): 8565–72. Дои:10.1021 / ja00048a032.
- ^ ИЮПАК, Сборник химической терминологии, 2-е изд. («Золотая книга») (1997). Исправленная онлайн-версия: (2006–) "стерический изотопный эффект ". Дои:10.1351 / goldbook.S06001
- ^ Mislow K, Грейв Р., Гордон А.Дж., Уол Г.Х. (1963). «Заметка о стерических изотопных эффектах. Конформационные кинетические изотопные эффекты в рацемизации 9,10-дигидро-4,5-диметилфенантрена». Журнал Американского химического общества. 85 (8): 1199–1200. Дои:10.1021 / ja00891a038.
- ^ Бартелл Л.С. (1 сентября 1961 г.). «Роль несвязанных отталкиваний во вторичных изотопных эффектах. I. Альфа и бета эффекты замещения.1». Журнал Американского химического общества. 83 (17): 3567–3571. Дои:10.1021 / ja01478a006.
- ^ Фелдер Т., Шалли, Калифорния (май 2003 г.). «Вторичные изотопные эффекты на реакцию удаления ротаксанов: высокоточное измерение стерического размера». Angewandte Chemie. 42 (20): 2258–60. Дои:10.1002 / anie.200350903. PMID 12772156.
- ^ Fleming DG, Arseneau DJ, Sukhorukov O, Brewer JH, Mielke SL, Schatz GC, Garrett BC, Peterson KA, Truhlar DG (январь 2011 г.). «Кинетические изотопные эффекты для реакций мюонного гелия и мюония с H2». Наука. 331 (6016): 448–50. Bibcode:2011Наука ... 331..448F. Дои:10.1126 / science.1199421. PMID 21273484.
- ^ Виберг КБ, Слау Л.Х. (1958). "Эффект изотопа дейтерия в галогенировании боковой цепи толуола". Журнал Американского химического общества. 80 (12): 3033–3039. Дои:10.1021 / ja01545a034.
- ^ Линч Р.А., Винченти С.П., Лин Ю.Т., Смакер Л.Д., Субба Рао С.К. (1972). «Аномальные кинетические эффекты изотопа водорода на скорость ионизации некоторых диалкилзамещенных кетонов». Журнал Американского химического общества. 94 (24): 8351–8356. Дои:10.1021 / ja00779a012.
- ^ Цзы В., Ван Ю. М., Тосте Ф. Д. (сентябрь 2014 г.). «Стратегия группы in situ для хирального анионного фторирования с переносом фазы в аллиловых спиртах». Журнал Американского химического общества. 136 (37): 12864–7. Дои:10.1021 / ja507468u. ЧВК 4183625. PMID 25203796.
дальнейшее чтение
- Белл Р.П., Крукс Дж. Э. (20 июля 1965 г.). «Кинетические эффекты изотопов водорода при ионизации некоторых кетоновых веществ». Труды Лондонского королевского общества A. 286 (1406): 285–299. Bibcode:1965RSPSA.286..285B. Дои:10.1098 / rspa.1965.0144.