Фармакокинетика - Википедия - Pharmacokinetics
Фармакокинетика (из Древнегреческий Pharmakon "наркотик" и кинетикос «движущийся, приводящий в движение»; видеть химическая кинетика ), иногда сокращенно ПК, это ветвь фармакология посвященный определению судьбы веществ, вводимых в живой организм. Интересующие вещества включают любые химические ксенобиотик Такие как: фармацевтические препараты, пестициды, пищевые добавки, косметика и др. Он пытается анализировать химические метаболизм и выяснить судьбу химического вещества с момента его введения до момента, когда оно полностью исключен из тела. Фармакокинетика - это изучение того, как организм влияет на лекарство, тогда как фармакодинамика (ПД) - это исследование того, как препарат влияет на организм. Оба вместе влияют дозирование, выгода, и побочные эффекты, как показано на Модели ПК / ПД.
- Процесс усвоения лекарств организмом, биотрансформация, которой они подвергаются, распределение лекарств и их метаболиты в тканях и выведение лекарств и их метаболитов из организма в течение определенного периода времени.
- Изучение других связанных процессов[1]
Обзор
Фармакокинетика описывает, как организм влияет на конкретный ксенобиотик / химическое вещество после введения через механизмы абсорбции и распределения, а также метаболические изменения вещества в организме (например, метаболические изменения). ферменты Такие как цитохром P450 или же глюкуронозилтрансфераза ферменты), а также эффекты и пути выведения метаболиты препарата.[2] Фармакокинетические свойства химических веществ зависят от способа введения и дозы введенного препарата. Это может повлиять на скорость всасывания.[3]
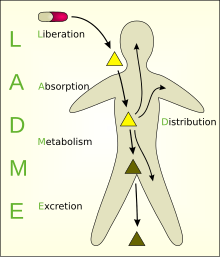
Модели были разработаны, чтобы упростить концептуальное представление многих процессов, которые происходят во взаимодействии между организмом и химическим веществом. Один из них, многокамерная модель, - наиболее часто используемые приближения к реальности; однако сложность добавления параметров при таком подходе моделирования означает, что одноквартирные модели и превыше всего две раздельные модели являются наиболее часто используемыми. Различные отсеки, на которые разделена модель, обычно называют ADME схема (также называемая LADME, если высвобождение включено в качестве отдельной стадии от абсорбции):
- Lосвобождение - процесс высвобождения лекарства из фармацевтический состав.[4][5] Смотрите также IVIVC.
- Апоглощение - процесс поступления вещества в кровоток.
- Dраспределение - рассеяние или распространение веществ по жидкостям и тканям тела.
- Mметаболизм (или биотрансформация, или инактивация) - признание организмом присутствия чужеродного вещества и необратимое преобразование исходных соединений в дочерние метаболиты.
- Excretion - выведение веществ из организма. В редких случаях некоторые наркотики безвозвратно накапливаются в ткани тела.[нужна цитата ]
Две фазы метаболизма и экскреции также можно сгруппировать под названием «устранение». Изучение этих отдельных фаз включает в себя использование основных понятий и манипулирование ими для понимания динамики процесса. По этой причине, чтобы полностью понять кинетика лекарственного средства необходимо детальное знание ряда факторов, таких как: свойства веществ, которые действуют как вспомогательные вещества, характеристики соответствующих биологические мембраны и способ, которым вещества могут пересекать их, или характеристики ферментативные реакции которые инактивируют препарат.
Все эти концепции можно представить через математические формулы которые имеют соответствующие графическое представление. Использование этих моделей позволяет понять характеристики молекула, а также то, как будет вести себя конкретный препарат, учитывая информацию о некоторых из его основных характеристик, таких как константа диссоциации кислоты (pKa), биодоступность и растворимость, абсорбционная способность и распределение в организме.
Результаты модели для лекарственного препарата можно использовать в промышленности (например, при расчете биоэквивалентность при разработке дженериков) или при клиническом применении фармакокинетических концепций. Клиническая фармакокинетика предоставляет множество руководств по эффективности для эффективного и действенного использования лекарств специалистами в области здравоохранения и Ветеринария.
Метрики
Ниже приведены наиболее часто измеряемые фармакокинетические показатели:[6] Единицы дозы в таблице выражены в родинки (моль) и коренной зуб (М). Чтобы выразить метрики таблицы в единицах массы, а не в Количество вещества просто замените "моль" на "г" и "М" на "г / дм3". Точно так же другие единицы в таблице могут быть выражены в единицах эквивалентной измерение масштабированием.
| Характеристика | Описание | Символ | Единица измерения | Формула | Пример работы ценить |
|---|---|---|---|---|---|
| Доза | Количество введенного препарата. | Расчетный параметр | 500 ммоль | ||
| Интервал дозирования | Время между введениями дозы препарата. | Расчетный параметр | 24 ч | ||
| CМаксимум | Пиковая концентрация препарата в плазме после приема. | Прямое измерение | 60,9 ммоль / л | ||
| тМаксимум | Время достигать CМаксимум. | Прямое измерение | 3.9 ч | ||
| Cмин | Нижайший (впадина ) концентрация, которой лекарство достигает до введения следующей дозы. | Прямое измерение | 27,7 ммоль / л | ||
| Объем распространения | Кажущийся объем, в котором распределено лекарство (т.е. параметр, связывающий концентрацию лекарства в плазме с количеством лекарства в организме). | 6.0 л | |||
| Концентрация | Количество препарата в данном объеме плазма. | 83,3 ммоль / л | |||
| Период полураспада | Время, необходимое для того, чтобы концентрация препарата удвоилась по сравнению с исходным значением для перорального и других внесосудистых путей введения.[нужна цитата ] | 1.0 ч | |||
| Константа скорости абсорбции | Скорость, с которой лекарство попадает в организм при пероральном и других внесосудистых путях. | 0.693 −1 | |||
| Период полувыведения | Время, необходимое для того, чтобы концентрация препарата достигла половины исходного значения. | 12 часов | |||
| Константа скорости удаления | Скорость, с которой лекарство выводится из организма. | 0,0578 ч−1 | |||
| Скорость инфузии | Скорость инфузии требуется для устранения баланса. | 50 ммоль / ч | |||
| Площадь под кривой | В интеграл кривой концентрация-время (после однократного приема или в стабильном состоянии). | 1320 ммоль / л · ч | |||
| Оформление | Объем плазмы, очищенной от препарата за единицу времени. | 0,38 л / ч | |||
| Биодоступность | Системно доступная фракция лекарственного средства. | Безразмерный | 0.8 | ||
| Колебания | Пиковые колебания минимума в пределах одного интервала дозирования в установившемся режиме. |
| 41.8% |







































В фармакокинетике устойчивое состояние относится к ситуации, когда общее потребление лекарства достаточно динамическое равновесие с его устранением. На практике обычно считается, что устойчивое состояние достигается, когда время, в 3-5 раз превышающее период полувыведения лекарственного средства после начала регулярного приема.
Фармакокинетические модели
Эта статья может потребоваться реорганизация для соответствия требованиям Википедии рекомендации по макету. (Апрель 2019) (Узнайте, как и когда удалить этот шаблон сообщения) |

Фармакокинетическое моделирование выполняется некоммерческими или разделенный методы. Некомпартментные методы оценивают воздействие лекарственного средства путем оценки площадь под кривой графика концентрация-время. Компартментные методы оценивают график концентрации-времени с использованием кинетических моделей. Некомпартментные методы часто более универсальны, поскольку они не предполагают какой-либо конкретной компартментальной модели и дают точные результаты, также приемлемые для исследований биоэквивалентности. Конечный результат превращений, которым лекарство подвергается в организме, и правила, которые определяют эту судьбу, зависят от числа взаимосвязанных факторов. Был разработан ряд функциональных моделей, чтобы упростить изучение фармакокинетики. Эти модели основаны на рассмотрении организма как ряда взаимосвязанных компонентов. Самая простая идея - думать об организме как об одном однородном компартменте. Этот одноквартирная модель предполагает, что плазма крови концентрации препарата являются истинным отражением концентрации препарата в других жидкостях или тканях, и что выведение препарата прямо пропорционально концентрации препарата в организме (кинетика первого порядка ).
Однако эти модели не всегда верно отражают реальную ситуацию в организме. Например, не все ткани тела одинаковы кровоснабжение, поэтому распределение препарата в этих тканях будет медленнее, чем в других с лучшим кровоснабжением. Кроме того, есть некоторые ткани (например, мозг ткани), которые представляют собой реальный барьер для распространения лекарств, который можно преодолеть с большей или меньшей легкостью в зависимости от характеристик лекарства. Если рассматривать эти относительные условия для различных типов тканей вместе со скоростью выведения, можно считать, что организм действует как два отдела: один, который мы можем назвать центральное отделение имеющий более быстрое распространение, включающий органы и системы с хорошо развитым кровоснабжением; и периферийный отсек состоит из органов с более низким кровотоком. Другие ткани, такие как мозг, могут занимать различное положение в зависимости от способности препарата проникать через барьер который отделяет орган от кровоснабжения.
Этот модель с двумя отсеками будет варьироваться в зависимости от того, в каком отсеке происходит удаление. Чаще всего выведение происходит в центральном отсеке, поскольку печень и почки органы с хорошим кровоснабжением. Однако в некоторых ситуациях может случиться так, что выведение происходит в периферическом отделении или даже в обоих. Это может означать, что существует три возможных варианта модели с двумя отсеками, которые еще не охватывают все возможности.[8]
Эта модель может быть неприменима в ситуациях, когда некоторые из ферментов, ответственных за метаболизм лекарства, становятся насыщенными, или когда присутствует активный механизм выведения, который не зависит от концентрации лекарства в плазме. В реальном мире каждая ткань будет иметь свои собственные характеристики распределения, и ни одна из них не будет строго линейной. Если мы обозначим препарат как объем распространения внутри организма VdF и его объем распределения в ткани VdТ Первый будет описан уравнением, которое учитывает все ткани, которые действуют по-разному, а именно:

Это представляет собой многокамерная модель с рядом кривых, которые выражают сложные уравнения, чтобы получить общую кривую. Номер компьютерные программы были разработаны для построения этих уравнений.[8] Какой бы сложной и точной ни была эта модель, она все же не соответствует действительности, несмотря на усилия, затраченные на получение различных значений распределения для лекарства. Это связано с тем, что понятие объема распределения является относительным понятием, которое не является истинным отражением реальности. Таким образом, выбор модели сводится к решению, какая из них предлагает наименьшую погрешность для рассматриваемого препарата.

Внекомпартментный анализ
Внекомпартментный PK-анализ сильно зависит от оценки общего воздействия лекарственного средства. Общее воздействие препарата чаще всего оценивается методом площади под кривой (AUC) с трапеция (численное интегрирование ) самый распространенный метод. Из-за зависимости от длины Икс в правиле трапеции оценка площади сильно зависит от графика отбора проб крови / плазмы. То есть, чем ближе точки времени, тем ближе трапеции отражают фактическую форму кривой зависимости концентрации от времени. Количество временных точек, доступных для выполнения успешного анализа NCA, должно быть достаточным, чтобы охватить фазы абсорбции, распределения и выведения, чтобы точно охарактеризовать лекарство. Помимо показателей воздействия AUC, с помощью методов NCA можно также сообщить такие параметры, как Cmax (максимальная концентрация), Tmax (время достижения максимальной концентрации), CL и Vd.
Компартментный анализ
Купе PK-анализ использует кинетические модели для описания и прогнозирования кривой зависимости концентрации от времени. Компартментные модели ПК часто похожи на кинетические модели, используемые в других научных дисциплинах, таких как химическая кинетика и термодинамика. Преимущество компартментного анализа перед некоторыми некомпартментными анализами заключается в возможности предсказать концентрацию в любое время. Недостатком является сложность разработки и проверки правильной модели. Моделирование без отсеков, основанное на разделении кривых, не страдает этим ограничением. Простейшей компартментной моделью PK является однокамерная модель PK с болюсным внутривенным введением и исключение первого порядка. Наиболее сложные модели ПК (называемые ПБПК модели) полагаются на использование физиологической информации для облегчения разработки и проверки.
Однокамерная модель
Линейная фармакокинетика называется так потому, что график взаимосвязи между различными вовлеченными факторами (доза, концентрации в плазме крови, выведение и т. д.) дает прямая линия или приближение к единице. Чтобы лекарства были эффективными, они должны иметь возможность быстро перемещаться из плазмы крови в другие жидкости и ткани организма.
Изменение концентрации с течением времени можно выразить как

Многосекционные модели

График нелинейной зависимости между различными факторами представлен в виде изгиб; Затем отношения между факторами могут быть найдены путем вычисления размеров различных областей под кривой. Модели, используемые в нелинейная фармакокинетика в значительной степени основаны на Кинетика Михаэлиса – Ментен К факторам нелинейности реакции относятся следующие:
- Многофазная абсорбция: вводимые препараты внутривенно удаляются из плазмы посредством двух основных механизмов: (1) распределение в тканях организма и (2) метаболизм + выведение лекарств. Результирующее снижение концентрации препарата в плазме крови происходит по двухфазной схеме (см. Рисунок).
 Концентрация препарата в плазме в зависимости от времени после в / в введения
Концентрация препарата в плазме в зависимости от времени после в / в введения- Альфа-фаза: начальная фаза быстрого снижения концентрации в плазме. Уменьшение в первую очередь связано с распределением лекарства из центрального отдела (циркуляции) в периферические отделы (ткани тела). Эта фаза заканчивается, когда между центральным и периферическим компартментами устанавливается псевдоравновесие концентрации лекарства.
- Бета-фаза: фаза постепенного снижения концентрации в плазме после альфа-фазы. Уменьшение в первую очередь связано с выведением лекарства, то есть его метаболизмом и выведением.[9]
- Иногда наблюдаются дополнительные фазы (гамма, дельта и т. Д.).[10]
- Характеристики препарата позволяют четко различать ткани с высоким и низким кровотоком.
- Ферментативный насыщенность: Когда доза препарата, выведение которого зависит от биотрансформации, увеличивается выше определенного порога, ферменты, ответственные за его метаболизм, становятся насыщенными. В этом случае концентрация препарата в плазме непропорционально возрастет, и его выведение не будет постоянным.
- Индукционная или ферментативное ингибирование: Некоторые препараты обладают способностью подавлять или стимулировать собственный метаболизм, в отрицательном или положительный отзыв реакции. Как это происходит с флувоксамин, флуоксетин и фенитоин. По мере введения больших доз этих фармацевтических препаратов концентрация неметаболизированного лекарственного средства в плазме увеличивается, и период полувыведения увеличивается. Поэтому необходимо скорректировать дозу или другие параметры лечения, когда требуется высокая доза.
- Почки также могут устанавливать механизмы активного выведения некоторых лекарств, независимо от их концентрации в плазме.

Таким образом, можно видеть, что нелинейность может возникать по причинам, которые влияют на всю фармакокинетическую последовательность: абсорбцию, распределение, метаболизм и выведение.
Биодоступность

На практическом уровне биодоступность лекарства можно определить как долю лекарства, которая достигает места своего действия. С этой точки зрения внутривенный введение лекарственного средства обеспечивает максимально возможную биодоступность, и считается, что этот метод дает биодоступность, равную 1 (или 100%). Биодоступность других методов доставки сравнивается с биодоступностью внутривенной инъекции (абсолютная биодоступность) или со стандартным значением, относящимся к другим методам доставки в конкретном исследовании (относительная биодоступность).
![{ displaystyle B_ {A} = { frac {[ABC] _ {P} cdot D_ {IV}} {[ABC] _ {IV} cdot D_ {P}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/e0349b27d143a975c5e5c3eeaf83b49e8c7a3318)
![{ displaystyle { mathit {B}} _ {R} = { frac {[ABC] _ {A} cdot { text {доза}} _ {B}} {[ABC] _ {B} cdot { text {доза}} _ {A}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/634ea7e9316eeae72a9904dcaf1ccb2c43511aaf)
После того, как биодоступность лекарственного средства установлена, можно рассчитать изменения, которые необходимо внести в его дозировку, чтобы достичь требуемых уровней в плазме крови. Следовательно, биодоступность - это математический фактор для каждого отдельного лекарства, который влияет на вводимую дозу. Можно рассчитать количество лекарственного средства в плазме крови, которое имеет реальный потенциал оказывать свое действие, используя формулу:

куда Де это эффективная доза, B биодоступность и Да введенная доза.
Следовательно, если лекарство имеет биодоступность 0,8 (или 80%) и его вводят в дозе 100 мг, уравнение покажет следующее:
- Де = 0,8 × 100 мг = 80 мг
То есть введенные 100 мг представляют собой концентрацию в плазме крови 80 мг, которая обладает способностью оказывать фармацевтический эффект.
Эта концепция зависит от ряда факторов, присущих каждому лекарству, таких как:[11]
- Форма выпуска
- Химическая форма
- Путь введения
- Стабильность
- Метаболизм
Эти концепции, которые подробно обсуждаются в соответствующих статьях, могут быть количественно определены математически и интегрированы для получения общего математического уравнения:

куда Q чистота препарата.[11]

куда скорость введения препарата и скорость, с которой абсорбированный препарат достигает кровеносной системы.

Наконец, используя Уравнение Хендерсона-Хассельбаха, и зная (pH при котором существует равновесие между его ионизированными и неионизированными молекулами), можно рассчитать неионизированную концентрацию лекарства и, следовательно, концентрацию, которая будет подвергаться абсорбции:


Когда два препарата имеют одинаковую биодоступность, они считаются биологическими эквивалентами или биоэквивалентами. Эта концепция биоэквивалентность важен, потому что в настоящее время он используется в качестве критерия при авторизации общие лекарства во многих странах.
LADME
После того, как лекарство вступает в контакт с организмом, происходит ряд фаз, которые описываются аббревиатурой LADME:
- Освобождение активного вещества из системы доставки,
- Абсорбция действующего вещества организмом,
- Распределение через плазму крови и различные ткани организма,
- Метаболизм то есть инактивация ксенобиотического вещества, и, наконец,
- Экскреция или же устранение вещества или продуктов его метаболизма.
Некоторые учебники объединяют первые две фазы, поскольку лекарство часто вводят в активной форме, что означает отсутствие фазы высвобождения. Другие включают фазу, которая объединяет распределение, метаболизм и выведение в фазу утилизации. Другие авторы включают токсикологический аспект препарата в так называемый ADME-Tox или же ADMET.
Каждая из фаз подвержена физико-химическим взаимодействиям между лекарством и организмом, которые можно выразить математически. Таким образом, фармакокинетика основана на математических уравнениях, которые позволяют прогнозировать поведение лекарственного средства и в которых большое внимание уделяется взаимосвязи между концентрацией лекарственного средства в плазме и временем, прошедшим с момента введения лекарственного средства.
Анализ
Биоаналитические методы
Биоаналитические методы необходимы для построения профиля концентрация-время. Химические методы используются для измерения концентрации наркотиков в биологическая матрица, чаще всего плазменный. Правильные биоаналитические методы должны быть избирательными и чувствительными. Например, микромасштабный термофорез может использоваться для количественной оценки того, как биологическая матрица / жидкость влияет на сродство лекарственного средства к его мишени.[12][13]
Масс-спектрометрии
Фармакокинетику часто изучают с помощью масс-спектрометрии из-за сложной природы матрикса (часто плазма или моча) и необходимости высокой чувствительности для наблюдения концентраций после низкой дозы и длительного периода времени. Чаще всего в этом приложении используется ЖХ-МС с тройной квадрупольный масс-спектрометр. Тандемная масс-спектрометрия обычно используется для дополнительной специфичности. Стандартные кривые и внутренние стандарты обычно используются для количественного определения одного фармацевтического препарата в образцах. Образцы представляют разные моменты времени, когда лекарство вводится, а затем метаболизируется или выводится из организма. Холостые пробы, взятые перед введением, важны для определения фона и обеспечения целостности данных с такими сложными матрицами проб. Большое внимание уделяется линейности стандартной кривой; однако обычно используют подгонка кривой с более сложными функциями, такими как квадратики поскольку отклик большинства масс-спектрометров не является линейным в больших диапазонах концентраций.[14][15][16]
В настоящее время существует значительный интерес к использованию масс-спектрометрии с очень высокой чувствительностью для микродозирование исследования, которые рассматриваются как многообещающая альтернатива эксперименты на животных.[17] Недавние исследования показывают, что Вторичная ионизация электрораспылением (SESI-MS) можно использовать для мониторинга лекарств, что дает преимущество в избежании жертвоприношения животных.[18]
Фармакокинетика населения
Фармакокинетика населения представляет собой исследование источников и коррелятов изменчивости концентраций лекарств среди лиц, которые являются целевой группой пациентов, получающих клинически значимые дозы интересующего лекарственного средства.[19][20][21] Определенные демографические, патофизиологические и терапевтические особенности пациента, такие как масса тела, выделительные и метаболические функции, а также наличие других методов лечения, могут регулярно изменять зависимости концентрации от дозы и могут объяснять вариабельность воздействия. Например, стационарные концентрации препаратов, выводимых в основном почками, обычно выше у пациентов, страдающих от почечная недостаточность чем у пациентов с нормальной функцией почек, получающих такую же дозу препарата. Популяционная фармакокинетика направлена на выявление поддающихся измерению патофизиологических факторов и объяснение источников вариабельности, которые вызывают изменения во взаимосвязи между дозой и концентрацией и степенью этих изменений, чтобы, если такие изменения связаны с клинически значимыми и значительными изменениями в экспозиции, которые влияют на терапевтический индекс доза может быть соответствующим образом изменена. Преимущество популяционного фармакокинетического моделирования заключается в его способности анализировать разреженные наборы данных (иногда доступно только одно измерение концентрации для каждого пациента).
Клиническая фармакокинетика
| Противоэпилептический медикамент | Кардиоактивный медикамент | Иммунодепрессор медикамент | Антибиотик медикамент |
|---|---|---|---|
| Бронходилататор медикамент | Цитостатический медикамент | Противовирусное средство (ВИЧ) лекарства | Факторы коагуляции |
| + Эфавиренц |
Клиническая фармакокинетика (возникающая в результате клинического использования фармакокинетики населения) - это прямое применение к терапевтической ситуации знаний о фармакокинетике лекарственного средства и характеристиках популяции, к которой принадлежит (или может быть отнесен) пациент.
Примером может служить возобновление использования циклоспорин как иммунодепрессор для облегчения трансплантации органов. Первоначально были продемонстрированы терапевтические свойства препарата, но после того, как выяснилось, что он вызывает нефротоксичность у ряда больных.[22] Однако затем стало понятно, что можно индивидуализировать дозу циклоспорина пациенту, анализируя плазматические концентрации пациентов (фармакокинетический мониторинг). Эта практика позволила снова использовать это лекарство и облегчить большое количество трансплантаций органов.
Клинический мониторинг обычно осуществляется путем определения концентраций в плазме, поскольку эти данные обычно наиболее легко получить и они наиболее надежны. К основным причинам определения концентрации препарата в плазме крови относятся:[23]
- Узкий терапевтический диапазон (разница между токсической и терапевтической концентрациями)
- Высокая токсичность
- Высокий риск для жизни.
Экотоксикология
Эта секция нуждается в расширении. Вы можете помочь добавляя к этому. (Апрель 2019) |
Экотоксикология это раздел науки, изучающий природу, эффекты и взаимодействие веществ, вредных для окружающей среды.[24][25]
Смотрите также
Рекомендации
- ^ Нордберг М., Даффус Дж., Темплтон Д.М. (1 января 2004 г.). «Глоссарий терминов, используемых в токсикокинетике (Рекомендации IUPAC 2003)». Чистая и прикладная химия. 76 (5): 1033–1082. Дои:10.1351 / pac200476051033. S2CID 98275795.
- ^ Фармакокинетика. (2006). В Словарь Мосби по медицине, медсестринскому делу и медицинским профессиям. Филадельфия, Пенсильвания: Elsevier Health Sciences. Получено 11 декабря 2008 г. из http://www.credoreference.com/entry/6686418
- ^ Рыцари К., Брайант Б. (2002). Фармакология для медицинских работников. Амстердам: Эльзевир. ISBN 0-7295-3664-5.
- ^ Кох HP, Ритчел В.А. (1986). «Освобождение». Synopsis der Biopharmazie und Pharmakokinetik (на немецком). Ландсберг, Мюнхен: Ecomed. С. 99–131. ISBN 3-609-64970-4.
- ^ Руис-Гарсия А., Бермехо М., Мосс А., Касабо В.Г. (февраль 2008 г.). «Фармакокинетика в открытии лекарств». Журнал фармацевтических наук. 97 (2): 654–90. Дои:10.1002 / jps.21009. PMID 17630642.
- ^ Рабочая группа AGAH PHARMACOKINETICS (2004-02-16). «Сборник терминов, символов, уравнений и объяснений общих фармакокинетических и фармакодинамических параметров и некоторых статистических функций» (PDF). Arbeitsgemeinschaft für Angewandte Humanpharmakologie (AGAH) (Ассоциация прикладной фармакологии человека). Архивировано из оригинал (PDF) на 2016-05-08. Получено 2011-04-04.
- ^ Интернет-ресурс по фармакокинетике Университет Лозанны Факультет биологии и медицины (FBM)
- ^ а б Майло Гибальди, Дональд Перье. FarmacocinéticaReverté 1982, страницы 1–10. ISBN 84-291-5535-Х, 9788429155358
- ^ Gill SC, Moon-Mcdermott L, Hunt TL, Deresinski S, Blaschke T., Sandhaus RA (сентябрь 1999 г.). «Фармакокинетика фазы I липосомального амикацина (MiKasome) у людей: дозозависимость и клиренс мочи». Abstr Intersci Conf Antimicrob Agents Chemother. 39: 33 (№ аннотации 1195).
- ^ Вайнер Д., Габриэльссон Дж. (2000). «ПК24 - Нелинейная кинетика - течение II». Фармакокинетический / фармакодинамический анализ данных: концепции и применение. Apotekarsocieteten. С. 527–36. ISBN 91-86274-92-9.
- ^ а б Майкл Э. Винтер, Мэри Энн Кода-Кимпл, Ллойд Ю. Янг, Эмилио Пол Янгуас Farmacocinética clínica básica Ediciones Díaz de Santos, 1994 стр. 8–14 ISBN 84-7978-147-5, 9788479781477 (на испанском языке)
- ^ Baaske P, Wienken CJ, Reineck P, Duhr S, Braun D (март 2010 г.). «Оптический термофорез для количественной оценки буферной зависимости связывания аптамера». Angewandte Chemie. 49 (12): 2238–41. Дои:10.1002 / anie.200903998. PMID 20186894. Сложить резюме – Phsyorg.com.
- ^ Винкен С.Дж., Баске П., Ротбауэр Ю., Браун Д., Дюр С. (октябрь 2010 г.). «Анализы связывания белков в биологических жидкостях с использованием термофореза на микроуровне». Nature Communications. 1 (7): 100. Bibcode:2010 НатКо ... 1..100 Вт. Дои:10.1038 / ncomms1093. PMID 20981028.
- ^ Ше Й., Корфмахер В.А. (июнь 2006 г.). «Повышение скорости и производительности при использовании систем ВЭЖХ-МС / МС для метаболизма лекарств и фармакокинетического скрининга». Текущий метаболизм лекарств. 7 (5): 479–89. Дои:10.2174/138920006777697963. PMID 16787157.
- ^ Кови Т.Р., Ли Э.Д., Хенион Д.Д. (октябрь 1986 г.). «Высокоскоростная жидкостная хроматография / тандемная масс-спектрометрия для определения лекарств в биологических образцах». Аналитическая химия. 58 (12): 2453–60. Дои:10.1021 / ac00125a022. PMID 3789400.
- ^ Кови Т. Р., Кроутер Дж. Б., Дьюи Е. А., Хенион Дж. Д. (февраль 1985 г.). «Термораспылительная жидкостная хроматография / масс-спектрометрическое определение лекарств и их метаболитов в биологических жидкостях». Аналитическая химия. 57 (2): 474–81. Дои:10.1021 / ac50001a036. PMID 3977076.
- ^ Комитет по лекарственным средствам для человека (CHMP) (декабрь 2009 г.). «Руководство ICH M3 (R2) по доклиническим исследованиям безопасности для проведения клинических испытаний на людях и регистрации фармацевтических препаратов» (PDF). Европейское агентство по лекарствам, Оценка лекарственных средств для человека. EMA / CPMP / ICH / 286/1995. Получено 4 мая 2013.
- ^ Ли, Сюэ; Мартинес-Лозано Синуес, Пабло; Даллманн, Роберт; Бреги, Лукас; Холлмен, Майя; Пру, Стивен; Браун, Стивен А .; Детмар, Майкл; Колер, Малькольм; Зеноби, Ренато (26.06.2015). «Фармакокинетика лекарств, определяемая анализом дыхания мышей в реальном времени». Angewandte Chemie International Edition. 54 (27): 7815–7818. Дои:10.1002 / anie.201503312. HDL:20.500.11850/102558. PMID 26015026.
- ^ Шейнер Л.Б., Розенберг Б., Марате В.В. (октябрь 1977 г.). «Оценка популяционных характеристик фармакокинетических параметров по рутинным клиническим данным». Журнал фармакокинетики и биофармацевтики. 5 (5): 445–79. Дои:10.1007 / BF01061728. PMID 925881. S2CID 28622472.
- ^ Шейнер Л. Б., Бил С., Розенберг Б., Марате В. В. (сентябрь 1979 г.). «Прогнозирование индивидуальной фармакокинетики». Клиническая фармакология и терапия. 26 (3): 294–305. Дои:10.1002 / cpt1979263294. PMID 466923. S2CID 41194071.
- ^ Бонате П.Л. (октябрь 2005 г.). «Рекомендуемая литература по популяционной фармакокинетической фармакодинамике». Журнал AAPS. 7 (2): E363–73. Дои:10.1208 / aapsj070237. ЧВК 2750974. PMID 16353916.
- ^ O'Valle, F .; García del Moral, R .; Андуджар, М. (1995). "Mecanismos de nefrotoxicidad por ciclosporina A a nivel celular". Нефрология (на испанском). 15 Приложение 1.
- ^ Хоакин Эррера Карранса Manual de farmacia clínica y Atención Farmacéutica (на испанском). Опубликовано Elsevier España, 2003; стр.159. ISBN 84-8174-658-4
- ^ Ягер Т., Альберт С., Пройс Т.Г., Ашауэр Р. (апрель 2011 г.). «Общая унифицированная пороговая модель выживания - токсикокинетико-токсикодинамическая основа экотоксикологии». Экологические науки и технологии. 45 (7): 2529–40. Bibcode:2011EnST ... 45.2529J. Дои:10.1021 / es103092a. PMID 21366215.
- ^ Ашауэр Р. «Токсикокинетико-токсикодинамические модели - экотоксикология и модели». Швейцарский федеральный институт водных наук и технологий. Архивировано из оригинал на 2012-04-05. Получено 2011-12-03.
внешняя ссылка
Эта статья использование внешняя ссылка может не следовать политикам или рекомендациям Википедии. (Май 2016) (Узнайте, как и когда удалить этот шаблон сообщения) |
Программного обеспечения
- Внеквартирный
- Бесплатное ПО: нести и ПК за р
- Коммерческий: MLAB, EquivTest, Кинетика, MATLAB / SimBiology, ПКМП,Феникс / WinNonlin, ПК Решения, RapidNCA.
- На базе отсека
- Бесплатное ПО: АДАПТ, Бумер (GUI ), SBPKPD.org (Фармакокинетика и фармакодинамика на основе системной биологии), WinSAAM, ПКфит для R, PharmaCalc и PharmaCalcCL, Java-приложения.
- Коммерческий: Ималитика, Kinetica, MATLAB / SimBiology, Феникс / WinNonlin, ПК Решения, Гончарный круг, ProcessDB, СААМ II.
- На физиологической основе
- Бесплатное ПО: MCSim
- Коммерческий: acslX, Cloe PK, GastroPlus, MATLAB / SimBiology, ПК-Сим, ProcessDB, Simcyp, Entelos PhysioLab Феникс / WinNonlin, Инструментальные средства ADME.
- Население ПК
- Бесплатное ПО: WinBUGS, АДАПТ, С-АДАПТ / САДАПТ-ТРАН, Бумер, PKBugs, Pmetrics для Р.
- Коммерческая: Kinetica, MATLAB / SimBiology, Моноликс, НЕМЕМ, Феникс / NLME, PopKinetics для SAAM II, USC * PACK, DoseMe-Rx, Верстак навигатора.
- Моделирование
Все программное обеспечение на основе модели, указанное выше.
- Бесплатное ПО: КОПАСИ, Мадонна Беркли, MEGen.
Образовательные центры
К глобальным центрам с самым высоким профилем предоставления углубленного обучения относятся университеты Буффало,Флорида, Гетеборг, Лейден, Отаго, Сан-Франциско, Пекин, Токио, Упсала, Вашингтон, Манчестер, Университет Монаша и Университет Шеффилд.[1]
| Авторитетный контроль |
|---|
- ^ Tucker GT (июнь 2012 г.). «Приоритеты исследований в фармакокинетике». Британский журнал клинической фармакологии. 73 (6): 924–6. Дои:10.1111 / j.1365-2125.2012.04238.x. ЧВК 3391520. PMID 22360418.