Физико-органическая химия - Physical organic chemistry
Эта статья поднимает множество проблем. Пожалуйста помоги Улучши это или обсудите эти вопросы на страница обсуждения. (Узнайте, как и когда удалить эти сообщения-шаблоны) (Узнайте, как и когда удалить этот шаблон сообщения)
|
Физико-органическая химия, термин, введенный Луи Хэммет в 1940 году относится к дисциплине органическая химия который фокусируется на отношениях между химические структуры и реактивность, в частности, с применением экспериментального инструментария физическая химия к изучению Органические молекулы. Конкретные основные направления исследования включают тарифы из органические реакции, Относительная химическая стабильность исходных материалов, реактивные промежуточные продукты, переходные состояния, и продукты химические реакции, и нековалентные аспекты сольватация и молекулярные взаимодействия которые влияют на химическую реактивность. Такие исследования предоставляют теоретические и практические основы для понимания того, как изменения структуры в растворах или твердотельных средах влияют на механизм реакции и ставка для каждого органическая реакция представляет интерес.
Заявление
Физико-органические химики используют теоретический и экспериментальные подходы помогают понять эти фундаментальные проблемы в органическая химия, в том числе классические и статистические термодинамический расчеты, квантово-механическая теория и вычислительная химия, а также экспериментальные спектроскопия (например., ЯМР ), спектрометрия (например., РС ), и кристаллография подходы. Таким образом, эта область имеет приложения к широкому спектру более специализированных областей, включая электро- и фотохимия, полимер и супрамолекулярная химия, и биоорганическая химия, энзимология, и химическая биология, а также коммерческим предприятиям, привлекающим технологическая химия, химическая инженерия, материаловедение и нанотехнологии, и фармакология в открытие лекарств по дизайну.
Объем
Эта секция требует дополнительных цитаты к вторичные или третичные источники (Июнь 2015 г.) (Узнайте, как и когда удалить этот шаблон сообщения) |
Физико-органическая химия исследование взаимосвязи между структурой и реактивностью Органические молекулы. В частности, физическая органическая химия применяет экспериментальные инструменты физическая химия к изучению структуры Органические молекулы и предоставляет теоретическую основу, которая интерпретирует, как структура влияет на оба механизмы и тарифы из органические реакции. Его можно рассматривать как подполе, соединяющее органическая химия с физическая химия.
Физико-органические химики используют как экспериментальные, так и теоретические дисциплины, такие как спектроскопия, спектрометрия, кристаллография, вычислительная химия, и квантовая теория изучить как тарифы из органические реакции и родственник химическая стабильность исходных материалов, переходные состояния, и продукты.[1][страница нужна ] Химики в этой области работают, чтобы понять физические основы современного органическая химия, поэтому физическая органическая химия находит применение в специализированных областях, включая химия полимеров, супрамолекулярная химия, электрохимия, и фотохимия.[1][страница нужна ]
История
Эта секция нуждается в расширении с: вдумчивым, кратким изложением важных вех в развитии этой специальной области химии. Вы можете помочь добавляя к этому. (Июнь 2015 г.) |
Период, термин физическая органическая химия сам был придуман Луи Хэммет в 1940 году, когда он использовал эту фразу в названии своего учебника.[2][нужна цитата ]
Химическая структура и термодинамика
Термохимия
Химики-органики используют инструменты термодинамика изучить связь, стабильность, и энергетика химических систем. Сюда входят эксперименты по измерению или определению энтальпия (ΔH), энтропия (ΔS) и Свободная энергия Гиббса (ΔG) реакции, превращения или изомеризации. Химики могут использовать различные химические и математические анализы, такие как Сюжет Ван 'т Хоффа, чтобы вычислить эти значения.
Эмпирические константы, такие как энергия диссоциации связи, стандартная теплота пласта (ΔHж°) и теплота сгорания (ΔHc°) используются для предсказания стабильности молекул и изменения энтальпия (ΔH) в ходе реакций. Для сложных молекул a ΔHж° значение может быть недоступно, но может быть оценено с использованием молекулярных фрагментов с известными высокая температура образования. Этот тип анализа часто называют Теория приращения группы Бенсона, в честь химика Сидни Бенсона, который посвятил всю свою карьеру разработке этой концепции.[1][страница нужна ] [3][4]
Термохимия реакционноспособных промежуточных продуктов -карбокатионы, карбанионы, и радикалы - также представляет интерес для физиков-органиков. Данные о групповом приращении доступны для радикальных систем.[1][страница нужна ] Карбокационную и карбанионную стабильность можно оценить с помощью сродства к гидрид-ионам и pKа значения, соответственно.[1][страница нужна ]
Конформационный анализ
Одним из основных методов оценки химической стабильности и энергетики является конформационный анализ. Физико-органические химики используют конформационный анализ для оценки различных типов напряжение присутствует в молекуле, чтобы предсказать продукты реакции.[5][страница нужна ] Штамм может быть обнаружен как в ациклических, так и в циклических молекулах, проявляясь в различных системах: скручивающая деформация, аллильный штамм, напряжение кольца, и син-пентановый штамм.[1][страница нужна ] A-ценности обеспечить количественную основу для прогнозирования конформация замещенного циклогексан, важный класс циклических органических соединений, реакционная способность которых во многом определяется конформационными эффектами. В Ценность разница в Свободная энергия Гиббса между осевой и экваториальной формами замещенного циклогексана, и путем сложения A-ценности различных заместители можно количественно предсказать предпочтительную конформацию производного циклогексана.
Помимо молекулярной стабильности, конформационный анализ используется для прогнозирования продуктов реакции. Один из часто цитируемых примеров использования конформационный анализ бимолекулярный реакция элиминации (E2). Эта реакция протекает наиболее легко, когда нуклеофил атакует вид, который антиперипланарный в уходящую группу. А молекулярная орбиталь Анализ этого явления показывает, что эта конформация обеспечивает лучшее перекрытие между электронами в R-H σ связывающая орбиталь подвергается нуклеофильной атаке, и пустой σ * разрушение орбитали разрываемой связи R-X.[6][страница нужна ] Используя этот эффект, конформационный анализ можно использовать для создания молекул, обладающих повышенной реакционной способностью.
Физические процессы, вызывающие барьеры вращения облигаций являются сложными, и эти барьеры широко изучены экспериментальными и теоретическими методами.[7][8][9] В ряде недавних статей исследовалось преобладание стерический, электростатический, и гиперконъюгативный вклад в вращательные барьеры в этан, бутан, и другие замещенные молекулы.[10]
Нековалентные взаимодействия

Химики используют исследование внутримолекулярных и межмолекулярных нековалентная связь / взаимодействия в молекулах для оценки реакционной способности. Такие взаимодействия включают, но не ограничиваются, водородная связь, электростатические взаимодействия между заряженными молекулами, диполь-дипольные взаимодействия, полярный-π и катион-π взаимодействия, π-укладка, донор-акцептор химия и галогенная связь. В дополнение гидрофобный эффект - ассоциация органических соединений в воде - электростатическая, нековалентное взаимодействие представляет интерес для химиков. Точное физическое происхождение гидрофобного эффекта происходит от многих сложные взаимодействия, но считается самым важным компонентом биомолекулярное распознавание в воде.[1][страница нужна ] Например, Сюй и Мельчер и другие. выяснил структурную основу распознавания фолиевой кислоты белками рецептора фолиевой кислоты.[11] Сильное взаимодействие между фолиевая кислота и рецептор фолиевой кислоты был приписан обоим водородные связи и гидрофобные взаимодействия. Изучение нековалентные взаимодействия также используется для изучения связывания и сотрудничество в супрамолекулярный сборки и макроциклические соединения Такие как краун-эфиры и криптанды, которые могут действовать как хозяева для гостевых молекул.
Кислотно-основная химия

Свойства кислоты и базы имеют отношение к физической органической химии. Химики-органики в первую очередь озабочены Бронстед – Лоури кислоты / основания в качестве доноров / акцепторов протонов и Кислоты / основания Льюиса как акцепторы / доноры электронов в органических реакциях. Химики используют ряд факторов, разработанных на основе физической химии: электроотрицательность /Индукция, прочность связи, резонанс, гибридизация, ароматичность, и сольватация - прогноз относительной кислотности и основности.
В принцип жесткой / мягкой кислоты / основания используется для предсказания молекулярных взаимодействий и направления реакции. Обычно предпочтительны взаимодействия между молекулами одного типа. То есть твердые кислоты будут ассоциироваться с твердыми основаниями, а мягкие кислоты - с мягкими. Концепция твердых кислот и оснований часто используется при синтезе неорганических координационные комплексы.
Кинетика
Физико-органические химики используют математические основы химической кинетики для изучения скорости реакций и механизмов реакций. В отличие от термодинамики, которая занимается относительной стабильностью продуктов и реагентов (ΔG °) и их равновесными концентрациями, изучение кинетики фокусируется на свободная энергия активации (ΔG‡) - разница в свободной энергии между структурой реагента и структурой переходного состояния - реакции, и, следовательно, позволяет химику изучать процесс уравновешивание.[1][страница нужна ] Математически полученные формализмы, такие как Постулат Хаммонда, то Принцип Куртина-Хэммета, а теория микроскопической обратимости часто применяются к органическая химия. Химики также использовали принцип термодинамический против кинетического контроля воздействовать на продукты реакции.
Законы о ставках
Изучение химическая кинетика используется для определения тарифный закон для реакции. Закон ставок устанавливает количественную зависимость между ставкой химическая реакция и концентрации или же давление присутствующих химических веществ.[12][страница нужна ] Законы скорости должны быть определены экспериментальным измерением и, как правило, не могут быть выяснены из химическое уравнение. Экспериментально определенный закон скорости относится к стехиометрии структура переходного состояния относительно структуры основного состояния. Исторически определение закона скорости осуществлялось путем мониторинга концентрации реагента во время реакции через гравиметрический анализ, но сегодня это делается почти исключительно с помощью быстрых и недвусмысленных спектроскопический техники. В большинстве случаев определение скоростных уравнений упрощается путем добавления большого избытка («затопление») всех реагентов, кроме одного.
Катализ
В этом разделе цитируется источники но не предоставляет ссылки на страницы. (Июнь 2015 г.) (Узнайте, как и когда удалить этот шаблон сообщения) |
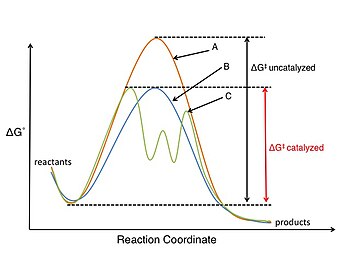
Изучение катализ и каталитические реакции очень важны для области физической органической химии. А катализатор участвует в химической реакции, но не расходуется в процессе.[12][страница нужна ] Катализатор снижает энергия активации барьер (ΔG‡), увеличивая скорость реакции либо за счет стабилизации структуры переходного состояния, либо за счет дестабилизации ключевого промежуточного продукта реакции, и, поскольку требуется лишь небольшое количество катализатора, он может обеспечить экономичный доступ к дорогостоящим или трудным для синтеза органическим молекулам. Катализаторы также могут влиять на скорость реакции, изменяя механизм реакции.[1][страница нужна ]
Кинетический изотопный эффект
Хотя закон скорости обеспечивает стехиометрию переходное состояние структура, она не предоставляет никакой информации о разрыве или образовании связей.[1][страница нужна ] Замена изотопа вблизи реактивного положения часто приводит к изменению скорости реакции. Изотопное замещение изменяет потенциальную энергию промежуточных продуктов реакции и переходных состояний, поскольку более тяжелые изотопы образуют более прочные связи с другими атомами. Атомная масса влияет на нулевую точку колебательное состояние связанных молекул, более короткие и более сильные связи в молекулах с более тяжелыми изотопами и более длинные и более слабые связи в молекулах с легкими изотопами.[6][страница нужна ] Поскольку колебательные движения часто меняются в ходе реакции из-за образования и разрыва связей, частоты будут затронуты, и замещение изотопа может дать представление о механизме реакции и законе скорости.
Заместительные эффекты
Изучение того, как заместители влияют на реакционную способность молекулы или скорость реакций, представляет значительный интерес для химиков. Заместители могут оказывать влияние как через стерический и электронные взаимодействия, последние из которых включают резонанс и индуктивные эффекты. В поляризуемость молекулы также могут быть затронуты. Большинство эффектов заместителей анализируются с помощью линейные отношения свободной энергии (LFER). Наиболее распространенным из них является Анализ графика Хэммета.[1][страница нужна ] В этом анализе сравнивается влияние различных заместителей на ионизацию бензойная кислота с их воздействием на различные химические системы. Параметры графиков Хаммета - сигма (σ) и rho (ρ). Значение σ указывает на кислотность замещенной бензойной кислоты по отношению к незамещенной форме. Положительное значение σ указывает на то, что соединение более кислое, а отрицательное значение указывает на то, что замещенный вариант менее кислый. Значение ρ является мерой чувствительности реакции к изменению заместителя, но измеряет только индукционные эффекты. Поэтому были созданы две новые шкалы, которые оценивают стабилизацию локализованного заряда через резонанс. Один - σ+, который касается заместителей, которые стабилизируют положительные заряды посредством резонанса, а другой - σ− который предназначен для групп, которые стабилизируют отрицательные заряды посредством резонанса. Анализ Хэммета может использоваться для выяснения возможных механизмов реакции. Например, если прогнозируется, что переходное состояние структура имеет накопление отрицательного заряда относительно структуры основного состояния, то электронодонорные группы можно ожидать увеличения скорости реакции.[1][страница нужна ]
Другой LFER весы были разработаны. Стерический и полярные эффекты анализируются через Параметры Taft. Замена растворителя вместо реагента может дать представление об изменениях заряда во время реакции. В График Грюнвальда-Винштейна дает количественное представление об этих эффектах.[1][страница нужна ] [13]
Эффекты растворителя
Эта секция слишком полагается на Рекомендации к основные источники. (Июнь 2015 г.) (Узнайте, как и когда удалить этот шаблон сообщения) |
Растворители может оказать сильное влияние на растворимость, стабильность, и скорость реакции. Смена растворителя также может позволить химику повлиять на термодинамический или кинетический контроль реакции. Реакции протекают с разной скоростью в разных растворителях из-за изменения распределения заряда во время химического превращения. Эффекты растворителя могут действовать в основном состоянии и / или переходное состояние конструкции.[1][страница нужна ]
Пример влияния растворителя на органические реакции можно увидеть на сравнение SN1 и SN2 реакции.[14][требуется дальнейшее объяснение ][пример необходим ]
Растворитель может также оказать значительное влияние на термодинамическое равновесие системы, например, как в случае кето-енольные таутомеризации. В неполярный апротический растворители, энол форма сильно предпочтительна из-за образования внутримолекулярного водородная связь, пока в полярный апротический растворители, такие как метиленхлорид, то энол форма менее предпочтительна из-за взаимодействия между полярным растворителем и полярным дикетон.[пример необходим ] В протический растворителей, равновесие находится в направлении кетоформы как внутримолекулярной водородная связь конкурирует с водородными связями, происходящими из растворителя.[15][неосновной источник необходим ] [16][неосновной источник необходим ] [17][неосновной источник необходим ]
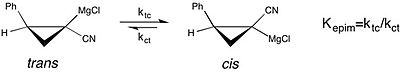
Современный пример изучения эффекты растворителя на химическое равновесие можно увидеть в исследовании эпимеризация из хиральный циклопропилнитрил Реактивы Гриньяра.[18][неосновной источник необходим ] Это исследование сообщает, что константа равновесия для СНГ к транс изомеризация из Реактив Гриньяра намного больше - предпочтение СНГ форма улучшена - в THF в качестве реакционного растворителя более диэтиловый эфир. Однако более высокая скорость цис-транс-изомеризация в THF приводит к потере стереохимический чистота. Это тот случай, когда понимание влияния растворителя на стабильность молекулярная конфигурация реагента важна с точки зрения селективности, наблюдаемой в асимметричный синтез.
Квантовая химия
Многие аспекты взаимосвязи структура-реакционная способность в органической химии можно рационализировать с помощью резонанс, толкание электронов, индукция, то правило восьми электронов, и s-p гибридизация, но это всего лишь полезные формализмы и не отражают физическую реальность. Из-за этих ограничений истинное понимание физической органической химии требует более строгого подхода, основанного на физика элементарных частиц. Квантовая химия обеспечивает строгую теоретическую основу, способную предсказывать свойства молекул путем расчета электронной структуры молекулы, и он стал легкодоступным инструментом для физиков-органиков в виде популярных пакетов программного обеспечения.[нужна цитата ] Сила квантовой химии основана на волновой модели атом, в которой ядро это очень маленькая, положительно заряженная сфера, окруженная диффузным электрон облако. Частицы определяются их связанными волновая функция, уравнение, которое содержит всю информацию, связанную с этой частицей.[12][страница нужна ] Вся информация о системе содержится в волновой функции. Эта информация извлекается из волновая функция за счет использования математических операторов.

Энергия, связанная с определенным волновая функция, возможно, самая важная информация, содержащаяся в волновой функции, может быть извлечена путем решения Уравнение Шредингера (вверху Ψ - волновая функция, E - энергия, Ĥ - гамильтонов оператор)[12][страница нужна ] в котором соответствующий Гамильтонов оператор применяется. В различных формах уравнения Шредингера общий размер распределения вероятностей частицы увеличивается с уменьшением массы частицы. По этой причине ядра имеют незначительный размер по сравнению с гораздо более легкими. электроны и рассматриваются как точечные заряды в практических приложениях квантовой химии.
Из-за сложных взаимодействий, которые возникают из-за отталкивания электронов, алгебраические решения уравнения Шредингера возможны только для систем с одним электроном, таких как водород атом, H2+, H32+, так далее.; однако из этих простых моделей возникают все знакомые атомный (s, p, d, f) и связывающие (σ, π) орбитали. В системах с множеством электронов общая многоэлектронная волновая функция описывает сразу все их свойства. Такие волновые функции генерируются путем линейного сложения волновых функций одного электрона для генерации первоначального предположения, которое многократно модифицируется, пока связанная с ним энергия не будет минимизирована. Чтобы найти удовлетворительное решение, часто требуются тысячи догадок, поэтому такие вычисления выполняются на мощных компьютерах. Важно отметить, что решения для атомов с множеством электронов придают такие свойства, как диаметр и электроотрицательность которые точно отражают экспериментальные данные и закономерности, обнаруженные в периодическая таблица. Растворы для молекул, таких как метан, дать точное представление об их электронная структура которые невозможно получить экспериментальными методами.[нужна цитата ] Вместо четырех дискретных σ-связей от углерода к каждому атому водорода теория предсказывает набор из четырех связывающих молекулярных орбиталей, которые делокализованы по всей молекуле. Точно так же истинная электронная структура 1,3-бутадиен показывает делокализованный π-соединение молекулярные орбитали протягивается через всю молекулу, а не через две изолированные двойные связи, как предсказывает простой Структура Льюиса.[нужна цитата ]
Полная электронная структура предлагает большую предсказательную силу для органических преобразований и динамики, особенно в случаях, касающихся ароматические молекулы, расширенный π системы, связи между ионами металлов и органическими молекулами, молекулы, содержащие нестандартные гетероатомы подобно селен и бор, а конформационная динамика больших молекул, таких как белки при этом многие приближения в химических формализмах делают невозможным предсказание структуры и реакционной способности. Примером того, как определение электронной структуры является полезным инструментом для химика-физика, является катализируемая металлом деароматизация бензол. Трикарбонил хрома очень электрофильный за счет изъятия электронной плотности из заполненных хром d-орбитали в разрушение CO орбитали, и может ковалентная связь к лицу молекулы бензола через делокализованный молекулярные орбитали. Сотрудничество лиганды индуктивно перенести электронную плотность из бензола через хром атом, и резко активировать бензол, чтобы нуклеофильный атака. Затем нуклеофилы могут реагировать с образованием гексациклодиенов, которые можно использовать в дальнейших превращениях, таких как Циклоаддитоны Diels Alder.[19]

Квантовая химия также может дать представление о механизме органического превращения без сбора каких-либо экспериментальных данных. Поскольку волновые функции обеспечивают полную энергию данного молекулярного состояния, предполагаемую молекулярную геометрию можно оптимизировать, чтобы получить расслабленные молекулярные структуры, очень похожие на те, которые были обнаружены с помощью экспериментальных методов.[20][страница нужна ] Координаты реакции затем можно смоделировать, и переходное состояние конструкции решены. Таким образом, возможно решение полной энергетической поверхности для данной реакции, и такие расчеты применялись для решения многих задач органической химии, где кинетические данные недоступны или их трудно получить.[1][страница нужна ]
Спектроскопия, спектрометрия и кристаллография
Физико-органическая химия часто влечет за собой идентификацию молекулярной структуры, динамики и концентрации реагентов в ходе реакции. Взаимодействие молекул со светом может дать огромное количество данных о таких свойствах посредством неразрушающего спектроскопические эксперименты, с свет поглощается, когда энергия фотона соответствует разнице в энергии между двумя состояния в молекуле и испускается, когда возбужденное состояние в молекуле коллапсирует до состояния с более низкой энергией. Спектроскопические методы широко классифицируются по типу исследуемого возбуждения, например: колебательный, вращающийся, электронный, ядерный магнитный резонанс (ЯМР) и электронный парамагнитный резонанс спектроскопия. Помимо спектроскопических данных, определению структуры часто помогают дополнительные данные, собранные из Дифракция рентгеновских лучей и масс-спектрометрический эксперименты.[21][страница нужна ]
ЯМР и ЭПР спектроскопия

Одним из самых мощных инструментов физической органической химии является ЯМР-спектроскопия. Внешний магнитное поле применяется к парамагнитный ядро генерирует два дискретных состояния, с положительным и отрицательным вращение значения расходятся энергия; затем можно определить разницу в энергии, определив частоту света, необходимую для возбуждения изменения состояния спина для данного магнитного поля. Ядра, которые нельзя различить в данной молекуле, поглощают на разных частотах, а общая площадь пика в спектре ЯМР пропорциональна количеству ядер, реагирующих на эту частоту.[22] Можно количественно оценить относительную концентрацию различных органических молекул, просто интеграция пики в спектре, и многие кинетические эксперименты могут быть легко и быстро выполнены, следя за развитием реакции в одном образце ЯМР. Протонный ЯМР часто используется синтетическими химиками-органиками, потому что протоны, связанные с определенными функциональные группы дают характерные энергии поглощения, но спектроскопию ЯМР можно проводить и на изотопы из азот, углерод, фтор, фосфор, бор, а множество других элементов. В дополнение к простым экспериментам по поглощению, также возможно определить скорость реакций обмена быстрыми атомами с помощью измерений обмена с подавлением, межатомные расстояния с помощью многомерных измерений. ядерный эффект оверхаузера экспериментов, а также спин-спиновая связь через гомоядерная корреляционная спектроскопия.[23] Помимо свойств спинового возбуждения ядер, можно также изучать свойства органических радикалы с помощью той же фундаментальной техники. У неспаренных электронов тоже есть сетка. вращение, а внешнее магнитное поле позволяет извлекать аналогичную информацию через электронный парамагнитный резонанс (ЭПР) спектроскопия.[1][страница нужна ]
Колебательная спектроскопия
Колебательная спектроскопия, или инфракрасная (ИК) спектроскопия, позволяет идентифицировать функциональные группы и, из-за его низкой стоимости и надежности, часто используется в учебных лабораториях и для мониторинга хода реакции в режиме реального времени в труднодоступных средах (высокое давление, высокая температура, газовая фаза, границы фаз ). Молекулярные колебания квантуются аналогично электронным волновым функциям, при этом целочисленное увеличение частоты приводит к более высокому энергетические состояния. Разница в энергии между колебательными состояниями почти постоянна, часто попадая в диапазон энергий, соответствующих инфракрасным фотонам, поскольку при нормальных температурах колебания молекул очень похожи на гармонические осцилляторы. Это позволяет грубо идентифицировать функциональные группы в Органические молекулы, но спектры усложняются колебательной связью между соседними функциональными группами в сложных молекулах. Поэтому его полезность при определении структуры обычно ограничивается простыми молекулами. Еще больше усложняет ситуацию то, что некоторые вибрации не вызывают изменения молекулярный дипольный момент и не будет наблюдаться с помощью стандартной ИК-спектроскопии поглощения. Вместо этого их можно исследовать через Рамановская спектроскопия, но этот метод требует более сложного оборудования и применяется реже. Однако, поскольку спектроскопия комбинационного рассеяния света основана на рассеянии света, ее можно проводить на микроскопических образцах, таких как поверхность гетерогенный катализатор, а фазовая граница или на подобразце объемом один микролитр (мкл) в большем объеме жидкости.[21][страница нужна ] Приложения колебательная спектроскопия часто используются астрономами для изучения состава облака молекулярного газа, внесолнечные планетные атмосферы, и планетарные поверхности.

Электронная спектроскопия возбуждения
Электронная спектроскопия возбуждения, или ультрафиолетовая-видимая (УФ-видимая) спектроскопия, выполняется в видимый и ультрафиолетовый регионы электромагнитный спектр и полезен для исследования разницы в энергии между самой высокой занятой энергией (HOMO) и самой низкой энергией незанятости (LUMO) молекулярные орбитали. Эта информация полезна физическим химикам-органикам при разработке органических фотохимический системы и красители, как поглощение разных длин волн видимый свет дайте Органические молекулы цвет. Поэтому подробное понимание электронной структуры полезно для объяснения электронных возбуждений, а посредством тщательного контроля молекулярной структуры можно настроить зазор HOMO-LUMO для получения желаемых цветов и свойств возбужденного состояния.[24]
Масс-спектрометрии
Масс-спектрометрии это метод, который позволяет измерять молекулярная масса и предлагает дополнительные данные для спектроскопический методы структурной идентификации. В типичном эксперименте образец газовой фазы органический материал является ионизированный и в результате ионные частицы ускоряются прикладным электрическое поле в магнитное поле. Отклонение, создаваемое магнитным полем, часто в сочетании со временем, которое требуется молекуле, чтобы достичь детектора, затем используется для расчета масса молекулы. Часто в процессе выборки ионизация большие молекулы распадаются на части, и полученные данные показывают исходную массу и ряд меньших масс фрагментов; такая фрагментация может дать хорошее представление о последовательности белков и полимеров нуклеиновых кислот. Помимо массы молекулы и ее фрагментов, распределение изотопный вариант массы также могут быть определены и качественное присутствие определенных элементов идентифицировано благодаря их характерным природным свойствам. распределение изотопов. Отношение популяции массы фрагмента к популяции исходного иона можно сравнить с библиотекой эмпирических данных о фрагментации и сопоставить с известной молекулярной структурой.[25] Комбинированный газовая хроматография и масс-спектрометрия используется для качественной идентификации молекул и количественного измерения концентрации с большой точностью и точностью, а также широко используется для проверки небольших количеств биомолекул и запрещенных наркотиков в образцах крови. Для химиков-синтетиков-органиков это полезный инструмент для характеристики новых соединений и продуктов реакции.
Кристаллография
В отличие от спектроскопических методов, Рентгеновская кристаллография всегда позволяет однозначно определять структуру и обеспечивает точные валентные углы и длины, которые полностью недоступны при спектроскопии. Он часто используется в физической органической химии для получения абсолютного молекулярная конфигурация и является важным инструментом в улучшении синтеза чистого энантиомерный субстанция. Это также единственный способ определить положение и связь элементов, у которых отсутствует ЯМР активный ядро Такие как кислород. Действительно, до того, как в начале 20 века стали доступны методы определения структуры рентгеновских лучей, все органические структуры были полностью предположительными: четырехгранный углерод, например, было подтверждено только Кристальная структура из алмаз,[26] а делокализованная структура бензола подтверждена Кристальная структура из гексаметилбензол.[27] Пока кристаллография предоставляет химикам-органикам весьма удовлетворительные данные, это не повседневный метод в органической химии, потому что идеальный монокристалл целевого соединения необходимо выращивать. Только сложные молекулы, для которых данные ЯМР не могут быть однозначно интерпретированы, нуждаются в этой методике. В приведенном ниже примере структуру комплекса хозяин-гость было бы довольно сложно решить без монокристаллической структуры: на кристалле нет протонов. фуллерен, и без ковалентных связей между двумя половинами органического комплекса одна только спектроскопия не смогла доказать предполагаемую структуру.[нужна цитата ]

дальнейшее чтение
Общий
- Питер Аткинс и Хулио де Паула, 2006, "Физическая химия", 8-е изд., Нью-Йорк, Нью-Йорк, США: Macmillan, ISBN 0716787598, видеть [2], по состоянию на 21 июня 2015 г. [Например, см. стр. 422 для теоретико-группового / симметрийного описания атомные орбитали способствует связыванию в метане, CH4, и стр. 390f для оценки энергии связи π-электрона для 1,3-бутадиен методом Хюккеля.]
- Томас Х. Лоури и Кэтлин Шуэллер Ричардсон, 1987 г., Механизм и теория в органической химии, 3-е изд., Нью-Йорк, штат Нью-Йорк, США: Harper & Row, ISBN 0060440848, видеть [3], по состоянию на 20 июня 2015 г. [Авторитетный учебник по этой теме, содержащий ряд приложений, в которых представлены технические подробности теории молекулярных орбиталей, кинетических изотопных эффектов, теории переходных состояний и радикальной химии.]
- Эрик В. Анслин и Деннис А. Догерти, 2006 г., Современная физико-органическая химия, Саусалито, Калифорния: University Science Books, ISBN 1891389319. [Модернизированный и оптимизированный подход с упором на приложения и междисциплинарные связи.]
- Майкл Б. Смит и Джерри, март, 2007 г., «Марш продвинутая органическая химия: реакции, механизмы и структура», 6-е изд., Нью-Йорк, Нью-Йорк, США: Wiley & Sons, ISBN 0470084944, видеть [4], по состоянию на 19 июня 2015 г.
- Фрэнсис А. Кэри и Ричард Дж. Сандберг, 2006, "Продвинутая органическая химия: Часть A: Структура и механизмы", 4-е изд., Нью-Йорк, Нью-Йорк, США: Springer Science & Business Media, ISBN 0306468565, видеть [5], по состоянию на 19 июня 2015 г.
История
- Хаммонд, Джордж С. (1997). «Физико-органическая химия через 50 лет: она изменилась, но сохранилась ли она?» (PDF). Pure Appl. Chem. 69 (9): 1919–22. Дои:10.1351 / pac199769091919. S2CID 53723796. Получено 20 июн 2015. [Выдающаяся отправная точка в истории области, от критически важного автора, ссылки и обсуждения раннего текста Хэммета и т. Д.]
Термохимия
- Л. К. Дорайсвами, 2005, «Оценка свойств органических соединений (гл. 3)», стр. 36–51, 118–124 (ссылки), в Инженерия органического синтеза, Оксфорд, Оксон, ENG: Oxford University Press, ISBN 0198025696, видеть [6], по состоянию на 22 июня 2015 г. (В этой главе книги рассматривается очень широкий спектр физических свойств и их оценка, включая узкий список термохимических свойств, приведенный в статье WP от июня 2015 г., в которой метод Бенсона и др. помещается рядом со многими другими методами. LK Дорайсвами Энсон Марстон, заслуженный профессор инженерных наук в Государственный университет Айовы.)
- Ирикура, Карл К .; Фрурип, Дэвид Дж. (1998). «Вычислительная термохимия». В Ирикуре Карл К .; Фрурип, Дэвид Дж. (Ред.). Вычислительная термохимия: прогноз и оценка молекулярной термодинамики. Серия симпозиумов ACS. 677. Американское химическое общество. С. 2–18. Дои:10.1021 / bk-1998-0677.ch001. ISBN 978-0-8412-3533-5.
Смотрите также
- Журнал физической органической химии
- Gaussian, пример используемого коммерчески доступного пакета квантово-механического программного обеспечения. особенно в академической среде
Рекомендации
- ^ а б c d е ж грамм час я j k л м п о п Догерти, Деннис А .; Анслин, Эрик В. (2006). Современная физико-органическая химия. Саусалито, Калифорния, США: Университетские научные книги. ISBN 9781891389313.[страница нужна ]
- ^ Хэммет, Луи П. (1940) Физико-органическая химия, Нью-Йорк, Нью-Йорк, США: Макгроу Хилл, см. [1], по состоянию на 20 июня 2015 г.
- ^ Cohen, N .; Бенсон, С. У. (1 ноября 1993 г.). «Оценка теплоты образования органических соединений методами аддитивности». Химические обзоры. 93 (7): 2419–2438. Дои:10.1021 / cr00023a005.
- ^ Бенсон, Сидней В .; Cruickshank, F. R .; Golden, D. M .; Haugen, Gilbert R .; O'Neal, H.E .; Роджерс, А. С .; Шоу, Роберт; Уолш Р. (1 июня 1969 г.). «Правила аддитивности для оценки термохимических свойств». Химические обзоры. 69 (3): 279–324. Дои:10.1021 / cr60259a002.
- ^ Кэри, Фрэнсис А. (2008). Органическая химия (7-е изд.). Бостон, Массачусетс, США: McGraw-Hill. ISBN 9780073047874.[страница нужна ]
- ^ а б Айзекс, Нил С. (1995). Физико-органическая химия (2-е изд.). Харлоу, ESS, ENG: Longman Scientific & Technical. ISBN 978-0582218635.[страница нужна ]
- ^ Мо, Иронг; Гао, Цзяли (1 февраля 2007 г.). «Теоретический анализ вращательного барьера этана». Отчеты о химических исследованиях. 40 (2): 113–119. Дои:10.1021 / ar068073w. PMID 17309192. S2CID 16332261.
- ^ Лю, Шубин (7 февраля 2013 г.). «Происхождение и природа барьеров для вращения облигаций: единый взгляд». Журнал физической химии A. 117 (5): 962–965. Bibcode:2013JPCA..117..962L. Дои:10.1021 / jp312521z. PMID 23327680.
- ^ Лю, Шубин; Говинд, Ниранджан (1 июля 2008 г.). «К пониманию природы барьеров внутреннего вращения с новой схемой разделения энергии: этан и бутан». Журнал физической химии A. 112 (29): 6690–6699. Bibcode:2008JPCA..112.6690L. Дои:10.1021 / jp800376a. PMID 18563887.
- ^ Ямамото, Такухей; Чен, Пи-Ю; Линь, Гуансинь; Блох-Мечкур, Анна; Jacobsen, Neil E .; Балли, Томас; Гласс, Ричард С. (1 октября 2012 г.). «Барьеры синтеза и вращения в 2,6-ди - (- анизил) анизоле» (PDF). Журнал физической органической химии. 25 (10): 878–882. Дои:10.1002 / poc.2939.
- ^ Чен, Чен; Кэ, Цзиюань; Чжоу, X. Эдвард; Йи, Вэй; Brunzelle, Joseph S .; Ли, Цзюнь; Йонг, Ю-Леонг; Сюй, Х. Эрик; Мелчер, Карстен (14 июля 2013 г.). «Структурные основы молекулярного распознавания фолиевой кислоты рецепторами фолиевой кислоты». Природа. 500 (7463): 486–489. Bibcode:2013Натура. 500..486C. Дои:10.1038 / природа12327. ЧВК 5797940. PMID 23851396.
- ^ а б c d McQuarrie, Donald A .; Саймон, Джон Д. (1997). Физическая химия: молекулярный подход (Ред. Ред.). Саусалито, Калифорния, США: Университетские научные книги. ISBN 9780935702996. Получено 21 июн 2015. Обратите внимание: Amazon, а не Google разрешает доступ к этому тексту.[страница нужна ]
- ^ Кевилл, Деннис Н .; Д'Суза, Малкольм Дж. (1 июня 1992 г.). «О разработке шкал ионизирующей способности растворителей на основе сольволизов бензильных субстратов». Журнал физической органической химии. 5 (6): 287–294. Дои:10.1002 / poc.610050602.
- ^ Райхард, Кристиан; Велтон, Томас (2011). Растворители и эффекты растворителей в органической химии (4-е, доп. И доп. Ред.). Вайнхайм, Германия: Wiley-VCH. ISBN 978-3-527-32473-6.
- ^ Миллс, Сандер Дж .; Клюв, Питер (1 апреля 1985 г.). «Влияние растворителей на кето-енольные равновесия: тесты количественных моделей». Журнал органической химии. 50 (8): 1216–1224. Дои:10.1021 / jo00208a014.[неосновной источник необходим ]
- ^ Эмсли, Джон; Фриман, Невилл Дж. (1 октября 1987 г.). «β-дикетонные взаимодействия». Журнал молекулярной структуры. 161 (1–2): 193–204. Bibcode:1987JMoSt.161..193E. Дои:10.1016/0022-2860(87)85074-3.[неосновной источник необходим ]
- ^ Шлунд, Себастьян; Basilio Janke, Eline M .; Вайс, Клаус; Энгельс, Бернд (1 января 2009 г.). «Предсказание таутомерного равновесия ацетилацетона в растворе. I. Правильный ответ по неправильной причине?». Журнал вычислительной химии. 31 (4): 665–70. Дои:10.1002 / jcc.21354. PMID 19557765. S2CID 6003410.[неосновной источник необходим ]
- ^ а б Гао, Мин; Patwardhan, Neeraj N .; Карлье, Пол Р. (2013). «Стереохимическая инверсия циано-стабилизированного реактива Гриньяра: замечательные эффекты эфирной структуры растворителя и концентрации». Варенье. Chem. Soc. 135 (38): 14390–14400. Дои:10.1021 / ja407348s. PMID 23978216.[неосновной источник необходим ]
- ^ Semmelhack, M. F .; Hall, H.T .; Йошифудзи, М. (сентябрь 1976 г.). «Промежуточные продукты .eta.5-циклогексадиенилтрикарбонилхрома (0) в реакции карбанионов с .eta.6-арентрикарбонилхромом (0)». Журнал Американского химического общества. 98 (20): 6387–6389. Дои:10.1021 / ja00436a056.
- ^ Шефер III, Генри Ф. (2004). Квантовая химия: развитие ab initio Методы теории электронного строения молекул.. Чикаго, Иллинойс, США: Р.Р.Доннелли (Курьер, Дувр). ISBN 978-0486432465. Получено 21 июн 2015.[страница нужна ]
- ^ а б Драго, Рассел С. (1992). Физические методы для химиков (2-е изд.). Ft. Уорт, Техас, США: Сондерс. ISBN 9780030970375. Получено 22 июн 2014.[страница нужна ]
- ^ Джеймс Килер. «ЯМР и уровни энергии (гл.2)» (PDF). Понимание ЯМР-спектроскопии. Калифорнийский университет в Ирвине. Получено 2013-10-26.
- ^ Киллер, Джеймс (2010). Понимание спектроскопии ЯМР (2-е изд.). Чичестер: Вайли. ISBN 978-0-470-74608-0.
- ^ Reusch, Уильям. «Видимая и ультрафиолетовая спектроскопия». Веб-сайт Университета штата Мичиган. Университет штата Мичиган. Получено 26 октября 2013.
- ^ Адлард, под редакцией Алан Дж. Хэндли, Эдвард Р. (2000). Методы и приложения газовой хроматографии. Бока-Ратон, Флорида: CRC Press. п. 168. ISBN 978-0-8493-0514-6.
- ^ Bragg, W. H .; Брэгг, В. Л. (июль 1913 г.). «Структура алмаза». Природа. 91 (2283): 557. Bibcode:1913Натура..91..557Б. Дои:10.1038 / 091557a0. S2CID 3987932.
- ^ Лонсдейл, К. (ноябрь 1928 г.). «Строение бензольного кольца». Природа. 122 (3082): 810. Bibcode:1928Натура.122..810Л. Дои:10.1038 / 122810c0. S2CID 4105837.
Филиалы химия | |
|---|---|
| Физический | |
| Органический | |
| Неорганический | |
| Аналитический | |
| Другие | |
| Смотрите также | |
| |