Лизосомная болезнь накопления - Википедия - Lysosomal storage disease
| Лизосомная болезнь накопления | |
|---|---|
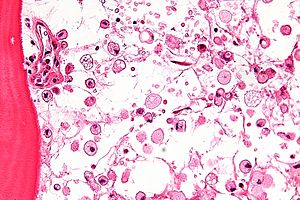 | |
| Микрофотография из Болезнь Гоше, с ячейками, имеющими характерный мятый бумажные салфетки -подобно цитоплазма. H&E пятно. | |
| Специальность | Эндокринология |
Лизосомные болезни накопления (LSD; /ˌлаɪsəˈsoʊмəl/) представляют собой группу из около 50 редких наследуемых метаболические нарушения которые возникают в результате нарушения функции лизосом.[1] Лизосомы представляют собой мешочки ферментов внутри клеток, которые переваривают большие молекулы и передают фрагменты другим частям клетки для повторного использования. Для этого процесса требуется несколько критических ферментов. Если один из этих ферментов неисправен из-за мутации, большие молекулы накапливаются внутри клетки, в конечном итоге убивая ее.[2]
Нарушения лизосомального накопления вызваны дисфункцией лизосом, как правило, в результате дефицита одного фермента, необходимого для метаболизм из липиды, гликопротеины (сахаросодержащие белки), или так называемые мукополисахариды. По отдельности ЛСД встречаются с частотой менее 1: 100 000; однако, как группа, заболеваемость составляет примерно 1: 5 000 - 1: 10 000.[3][4] Большинство этих расстройств аутосомно-рецессивно унаследовано, например, Болезнь Ниманна – Пика, тип C, но некоторые из них Х-сцепленный рецессивно унаследовано, например Болезнь Фабри и Синдром Хантера (MPS II).
Лизосому обычно называют центром переработки клеток, потому что она перерабатывает нежелательный материал в вещества, которые клетка может использовать. Лизосомы разрушают это нежелательное вещество за счет ферменты, узкоспециализированный белки необходим для выживания. Лизосомные расстройства обычно возникают, когда определенный фермент присутствует в слишком малом количестве или отсутствует совсем. Когда это происходит, в клетке накапливаются вещества. Другими словами, когда лизосома не функционирует нормально, избыточные продукты, предназначенные для разложения и повторного использования, хранятся в клетке.
Как и другие генетические нарушения, люди наследуют лизосомные болезни накопления от своих родителей. Хотя каждое заболевание возникает в результате различных генных мутаций, которые приводят к дефициту активности ферментов, все они имеют общую биохимическую характеристику - все лизосомные расстройства возникают из-за аномального накопления веществ внутри лизосомы.
ЛСД поражают в основном детей, и они часто умирают в молодом возрасте, многие в течение нескольких месяцев или лет после рождения.
Классификация
Стандартная классификация
LSD обычно классифицируются по природе первичного хранимого материала и в целом могут быть разбиты на следующие: (МКБ-10 коды предоставляются там, где они доступны)
- (E75) Нарушения накопления липидов
- Сфинголипидозы, включая Гоше и Болезни Ниманна – Пика (E75.0-E75.1)
- Ганглиозидоз (включая Болезнь Тея – Сакса (E75.2)
- Лейкодистрофии
- (E76.0) Мукополисахаридозы, включая Синдром Хантера и Болезнь Гурлера
- (E77) Нарушения накопления гликопротеинов
- (E77.0-E77.1) Муколипидозы
Также, болезнь накопления гликогена типа II (Болезнь Помпе) также является дефектом лизосомального метаболизма,[5] хотя в остальном он классифицируется как E74.0 в МКБ-10. Цистиноз ЛСД, характеризующийся аномальным накоплением аминокислоты цистина.
По типу дефектного белка
В качестве альтернативы белкам-мишеням LSD можно классифицировать по типу белка, который является дефицитным и вызывает накопление.
| Тип дефектного белка | Примеры болезней | Недостаток белка |
|---|---|---|
| Лизосомальный ферменты в первую очередь | Болезнь Тея – Сакса, I-клеточная болезнь,[6] Сфинголипидозы (например., Болезнь Краббе, ганглиозидоз: Гоше, Болезнь Ниманна – Пика и гликолипиды: Метахроматическая лейкодистрофия ), Дефицит липазы лизосомальной кислоты | Разные |
| Посттрансляционная модификация ферментов | Множественный дефицит сульфатазы | Множественные сульфатазы |
| Мембранные транспортные белки | Муколипидоз тип II и IIIA | N-ацетилглюкозамин-1-фосфаттрансфераза |
| Белки, защищающие ферменты | Галактосиалидоз | Катепсин А |
| Растворимые неферментативные белки | Дефицит GM2-AP, вариант AB, Болезнь Ниманна – Пика, тип C2 | GM2-AP, NPC2 |
| Трансмембранные белки | Дефицит SAP | Белки-активаторы сфинголипидов |
| Болезнь Ниманна – Пика, тип С1 | NPC1 | |
| Болезнь Саллы | Сиалин | |
| Если иное не указано в полях, применимая ссылка:[7] | ||
Лизосомные нарушения накопления
Это ЛСД:
- Сфинголипидозы
- Церамидаза
- Болезнь Фарбера
- Болезнь Краббе
- Инфантильное начало
- Позднее начало
- Галактосиалидоз
- Ганглиозиды: ганглиозидозы
- Альфа-галактозидаза
- Болезнь Фабри (альфа-галактозидаза А)
- Болезнь Шиндлера (альфа-галактозидаза B)
- Бета-галактозидаза / Ганглиозидоз GM1
- Инфантильный
- Несовершеннолетний
- Взрослый / хронический
- GM2 ганглиозидоз
- Вариант AB
- Недостаток активатора
- Болезнь Сандхоффа
- Инфантильный
- Несовершеннолетний
- Начало у взрослых
- Тай – Сакс
- Ювенильный дефицит гексозаминидазы А
- Хронический дефицит гексозаминидазы А
- Альфа-галактозидаза
- Глюкоцереброзид
- Болезнь Гоше
- Тип I
- Тип II
- Тип III
- Болезнь Гоше
- Сфингомиелиназа
- Дефицит липазы лизосомальной кислоты
- Раннее начало
- Позднее начало
- Болезнь Ниманна – Пика
- Введите
- Тип B
- Дефицит липазы лизосомальной кислоты
- Сульфатидоз
- Метахроматическая лейкодистрофия
- Дефицит сапозина B
- Множественный дефицит сульфатазы
- Метахроматическая лейкодистрофия
- Тип I
- MPS I Синдром Гурлера
- MPS I S Синдром Шейе
- MPS I H-S Синдром Херлера – Шейе
- Тип II (Синдром Хантера )
- Тип III (Синдром Санфилиппо )
- MPS III A (тип A)
- MPS III B (тип B)
- MPS III C (тип C)
- MPS III D (тип D)
- Тип IV (Моркио )
- MPS IVA (тип A)
- MPS IVB (Тип B)
- Тип VI (Синдром Марото-Лами )
- Тип VII (Хитрый синдром )
- Тип IX (дефицит гиалуронидазы )
Муколипидоз
- Тип I (сиалидоз )
- Тип II (I-клеточная болезнь )
- Тип III (псевдогурлер-полидистрофия / фосфотрансфераза дефицит)
- Тип IV (дефицит муколипидина 1 )
- Болезнь Ниманна – Пика
- тип C
- Тип D
- Нейрональные цероидные липофусцинозы
- Тип 1 Болезнь Сантавуори – Халтии / инфантильный NCL (CLN1 PPT1 )
- Тип 2 Болезнь Янского – Бельшовского / поздний младенец NCL (CLN2 / LINCL TPP1 )
- Тип 3 Болезнь Баттена – Шпильмейера – Фогта / ювенильный NCL (CLN3 )
- Тип 4 Болезнь Куфа / взрослый NCL (CLN4 )
- Тип 5 Финский вариант / поздний младенец (CLN5 )
- Тип 6 Поздний инфантильный вариант (CLN6 )
- Тип 7 CLN7
- Северная эпилепсия 8 типа (CLN8 )
- Турецкий поздний инфантильный тип 8 (CLN8 )
- Немецкий / сербский поздний младенческий тип 9 (неизвестно)
- Тип 10 Врожденный дефицит катепсина D (CTSD )
- Болезнь Вольмана
Лизосомальные транспортные болезни
- Цистиноз
- Пикнодизостоз
- Болезнь Саллы / болезнь накопления сиаловой кислоты
- Детская болезнь накопления свободной сиаловой кислоты
Болезни накопления гликогена
- Тип II Болезнь Помпе
- Тип IIb Болезнь Данона [8]
Другой
Лизосомная болезнь
Признаки и симптомы
Симптомы ЛСД варьируются в зависимости от конкретного расстройства и других переменных, таких как возраст начала, и могут быть от легких до тяжелых. Они могут включать задержку развития, двигательные нарушения, припадки, слабоумие, глухота, и / или слепота. У некоторых людей с ЛСД есть увеличенная печень или же селезенка, легочный и сердечный проблемы и кости, которые ненормально растут.[9]
Диагностика
Большинство пациентов сначала проходят скрининг с помощью ферментного анализа, который является наиболее эффективным методом для постановки окончательного диагноза.[9] В некоторых семьях, где известны мутации, вызывающие заболевание, и в некоторых генетических изолятах может быть проведен анализ мутаций. Кроме того, после того, как диагноз установлен биохимическим методом, для некоторых нарушений может быть проведен анализ мутаций.
Уход
Лекарства от лизосомальных болезней накопления не известны, и лечение в основном симптоматическое, хотя трансплантация костного мозга и заместительная ферментная терапия (ERT) были опробованы с некоторым успехом.[10][11] ФЗТ может минимизировать симптомы и предотвратить необратимое повреждение организма.[12] Кроме того, пуповинная кровь Трансплантация проводится в специализированных центрах по лечению ряда этих заболеваний. Кроме того, субстратная терапия, метод, используемый для уменьшения производства материала для хранения, в настоящее время оценивается для некоторых из этих заболеваний. Более того, шаперонная терапия метод, используемый для стабилизации дефектных ферментов, производимых пациентами, исследуется на предмет некоторых из этих нарушений. Экспериментальная методика генная терапия может предложить лекарства в будущем.[13]
Амброксол недавно было показано, что он увеличивает активность лизосомального фермента глюкоцереброзидазы, поэтому он может быть полезным терапевтическим средством как при болезни Гоше, так и при болезнь Паркинсона.[14][15] Амброксол вызывает секрецию лизосомы из клеток путем индукции рН-зависимой высвобождение кальция из кислых запасов кальция.[16] Следовательно, освобождение клетки от накопления продуктов разложения - это предложенный механизм, с помощью которого может помочь это лекарство.
История
Болезнь Тея – Сакса был первым из этих расстройств, описанных в 1881 г., за которым последовали Болезнь Гоше в 1882 году. В конце 1950-х - начале 1960-х годов де Дюв и его коллеги, используя методы фракционирования клеток, цитологический исследования и биохимические анализы идентифицировали и охарактеризовали лизосому как клеточную органеллу, ответственную за внутриклеточный переваривание и переработка макромолекулы. Это был научный прорыв, который привел к пониманию физиологической основы ЛСД. Болезнь Помпе было первым заболеванием, которое было идентифицировано как ЛСД в 1963 году, при этом Л. Херс сообщил, что причиной этого является дефицит α-глюкозидазы. Она также предположила, что другие болезни, такие как мукополисахаридоз, может быть связано с недостаточностью ферментов.
Смотрите также
Рекомендации
- ^ Винчестер Б., Веллоди А., Янг Е. (2000). «Молекулярные основы лизосомальных болезней накопления и их лечение». Biochem. Soc. Транс. 28 (2): 150–4. Дои:10.1042 / bst0280150. PMID 10816117.
- ^ Рис, Джейн; Кэмпбелл, Нил (2002). Биология. Сан-Франциско: Бенджамин Каммингс. стр.121–122. ISBN 0-8053-6624-5.
- ^ Meikle, P.J .; Hopwood, J. J .; Clague, A.E .; Кэри, У. Ф. (20 января 1999 г.). «Распространенность лизосомных нарушений накопления». JAMA. 281 (3): 249–254. Дои:10.1001 / jama.281.3.249. ISSN 0098-7484. PMID 9918480.
- ^ М, Фуллер; PJ, Meikle; JJ, Хопвуд (1 января 2006 г.). «Эпидемиология лизосомальных болезней накопления: обзор». PMID 21290699. Цитировать журнал требует
| журнал =(помощь) - ^ eMedicine Specialities> Неврология> Детская неврология> Лизосомная болезнь накопления Автор: Ной С. Шейнфельд, доктор медицины, доктор медицинских наук, FAAD. Соавтор (и): Ровена Эмилия Табамо, доктор медицины; Брайан Кляйн, доктор медицины. Обновлено: 25 сентября 2008 г.
- ^ Медицинская физиология (2-е издание) - W. Boron & E. Boulpaep, Saunders Press
- ^ Таблица 7-6 в:Митчелл, Ричард Шеппард; Кумар, Винай; Аббас, Абул К .; Фаусто, Нельсон (2007). Базовая патология Роббинса. Филадельфия: Сондерс. ISBN 978-1-4160-2973-1. 8-е издание.
- ^ «Болезнь Данона».
- ^ Кларк Дж. Т., Иваночко Р. М. (2005). «Заместительная ферментная терапия болезни Фабри». Мол. Нейробиол. 32 (1): 043–050. Дои:10.1385 / МН: 32: 1: 043. PMID 16077182.
- ^ Бруни С., Лоши Л., Инсерти С., Габриелли О., Коппа Г. В. (2007). «Последняя информация о лечении лизосомных болезней накопления». Acta Myol. 26 (1): 87–92. ЧВК 2949325. PMID 17915580.
- ^ «Ферментно-заместительная терапия болезни Гоше». Национальный фонд Гоше. Получено 2017-06-08.
- ^ Вдумайтесь К.П., Хаскинс М.Э. (2007). «Генная терапия мукополисахаридоза». Мнение эксперта Biol Ther. 7 (9): 1333–1345. Дои:10.1517/14712598.7.9.1333. ЧВК 3340574. PMID 17727324.
- ^ Макнил, Алисдэр; Magalhaes, Joana; Шен, Чэнго; Чау, Кай-Инь; Хьюз, Дерралин; Мехта, Атул; Фолтыние, Том; Купер, Дж. Марк; Абрамов, Андрей Юрьевич (2014-05-01). «Амброксол улучшает биохимию лизосом в клетках болезни Паркинсона, связанных с мутацией глюкоцереброзидазы». Мозг. 137 (5): 1481–1495. Дои:10.1093 / мозг / awu020. ISSN 0006-8950. ЧВК 3999713. PMID 24574503.
- ^ Альбин, Роджер Л .; Дауэр, Уильям Т. (01.05.2014). "Волшебное ружье от болезни Паркинсона?". Мозг. 137 (5): 1274–1275. Дои:10.1093 / мозг / awu076. ISSN 0006-8950. PMID 24771397.
- ^ Фуа, Джорджио; Хоби, Нина; Фелдер, Эдвард; Зиглер, Андреас; Миклавц, Пика; Вальтер, Пол; Радермахер, Питер; Халлер, Томас; Дитль, Пол (2015). «Новая роль старого препарата: амброксол запускает лизосомный экзоцитоз за счет pH-зависимого высвобождения Ca2 + из кислых запасов Ca2 +». Клеточный кальций. 58 (6): 628–637. Дои:10.1016 / j.ceca.2015.10.002. PMID 26560688.
внешняя ссылка
| Классификация |
|---|