Синдром Нунана с множественными лентиго - Noonan syndrome with multiple lentigines
| Синдром Нунана с множественными лентиго (NSML) | |
|---|---|
| Другие имена | Синдром ЛЕОПАРДА, кожно-сердечный синдром, синдром Горлина II, синдром профузы лентигиноза, прогрессирующий кардиомиопатический лентигиноз,[1]:550 Синдром Капуте-Римоина-Кенигсмарка-Эстерли-Ричардсона, синдром Мойнахана |
 | |
| Три четверти лица, пациент в первом поколении демонстрирует легкую прогнатизм и низко посаженные уши | |
| Специальность | Медицинская генетика |
Синдром Нунана с множественными лентиго (NSML), которая является частью группы, называемой Рас /MAPK синдромы проводящих путей,[2] редкий аутосомно-доминантный,[3] мультисистемное заболевание, вызванное мутация в протеинтирозинфосфатаза, ген нерецепторного типа 11 (ПТПН11 ). Заболевание представляет собой комплекс признаков, в основном затрагивающих кожу, скелетную и сердечно-сосудистую системы, которые могут присутствовать или не присутствовать у всех пациентов. Природа того, как мутация вызывает каждый из симптомов заболевания, не очень хорошо известна; однако исследования продолжаются. Это РАСопатия.
Синдром Нунана с множественными лентиго вызвано другим миссенс-мутация того же гена. Синдром Нунана довольно часто (от 1: 1000 до 1: 2500 живорожденных), и нейрофиброматоз 1 (который когда-то считался связанным с NSML) также распространен (1: 3500); однако нет эпидемиологический данные существуют для NSML.[4]
Признаки и симптомы
Альтернативное название состояния - синдром ЛЕОПАРДА - это мнемонический, первоначально выпущенный в 1969 году,[5] поскольку это состояние характеризуется некоторыми из следующих семи условий, первые буквы которых означают ЛЕОПАРД вместе с характеристикой "веснушка "кожи, вызванной лентиго это напоминает большая кошка.
- Лентигины - от красновато-коричневого до темно-коричневого пятна (поверхность кожи поражение ) обычно встречается в большом количестве (10000+) на большой части кожи, иногда превышая 80%. Они могут появиться даже во рту (щечный ), либо на поверхности глаза (склеральный ). Они имеют неровные границы и имеют размер от 1 мм до кафе с молоком, несколько сантиметров в диаметре. Также некоторые области витилиго -подобно гипопигментация может наблюдаться.
- Электрокардиографический проводимость аномалии: Обычно наблюдается на электрокардиограф как межжелудочковая блокада.
- Окуляр гипертелоризм: Широко расставленные глаза, что приводит к схожему внешнему виду пациентов. Аномалии лица - второй по частоте встречающийся симптом после лентиго. К аномалиям также относятся: широкий корень носа, прогнатизм (выступающая нижняя челюсть) или низко посаженные, возможно повернутые уши.
- Легочный стеноз: Сужение легочная артерия как он выходит из сердце. Могут присутствовать другие сердечные аномалии, в том числе: стеноз аорты, или же пролапс митрального клапана.
- Аномальный гениталии: обычно крипторхизм (сохранение яички в теле) или монорхизм (одно яичко). У пациенток это проявляется в виде отсутствующих или единичных яичников, которые по природе гораздо труднее обнаружить. Ультразвуковое сканирование проводится через регулярные промежутки времени, начиная с возраста 1 года, чтобы определить наличие яичников.
- Замедленный рост: Медленный или задержка роста. Большинство новорожденных с этим синдромом имеют нормальный вес и рост при рождении, но часто в течение первого года их рост замедляется.
- Глухота: Нейросенсорная (нервная глухота).
Наличие всех этих клейма не нужен для диагностики. Клинический диагноз считается произведенным, когда с лентиго Присутствуют 2 других наблюдаемых симптома, такие как отклонения ЭКГ и глазной гипертелоризм, или без лентиго, присутствуют 3 из вышеуказанных состояний, у родственника первой степени родства (т.[6]
- Дополнительные дерматологические аномалии (подмышечные веснушки, локализованные гипопигментация, межпальцевые лямки, гиперупругая кожа)
- Легкая умственная отсталость наблюдается примерно у 30% людей, страдающих синдромом.
- Нистагм (непроизвольные движения глаз), припадки, или же гипосмия (снижение обоняния) было зарегистрировано у нескольких пациентов.
- В 2004 г. у пациента был зарегистрирован рецидив заболевания верхней конечности. аневризмы что потребовало хирургического ремонта.[7]
- В 2006 году у пациента с NSML было зарегистрировано острый миелолейкоз.[8]
Из-за редкости самого синдрома трудно определить, действительно ли некоторые дополнительные заболевания являются частью синдрома. При базовой популяции, возможно, менее тысячи человек, один или два крайних случая могут очень быстро исказить статистическую популяцию.

Рука 37-летнего пациента показывает межпальцевые лямки

Пациент 37 лет (второе поколение) с гипертелоризмом, широким корнем носа, легким птозом.
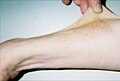
Пациент 37 лет демонстрирует гиперэластичность

21-месячный пациент в третьем поколении, подтвержденный генетическими тестами как Y279C, проявляющий глазной гипертелиоризм, цефало-лицевое сходство.

Торс 37-летнего пациента во втором поколении, демонстрирующего лентигиноз.




Патофизиология

В двух преобладающих мутациях NSML (Y279C и T468M ) мутации вызывают потерю каталитическая активность белка SHP2 (продукт гена ПТПН11 ген), что является ранее нераспознанным поведением для этого класса мутаций.[9] Это мешает фактору роста и связанной с ним передаче сигналов. Хотя дальнейшие исследования подтверждают этот механизм,[10][11] необходимы дополнительные исследования, чтобы определить, как это связано со всеми наблюдаемыми эффектами NSML.
Диагностика
Наличие заболевания можно подтвердить с помощью генетического теста. В исследовании 10 младенцев с клиническими показаниями к NSML до их первого дня рождения было подтверждено наличие подозреваемой мутации у 8 (80%) пациентов. Впоследствии было обнаружено, что еще у одного пациента с подозреваемой мутацией NF1, после оценки матери.[12]
Выявлено 5 аллельный варианты отвечает за NSML. Y279C, T468M, A461T, G464A, и Q510P что кажется уникальной семейной мутацией, поскольку все другие варианты вызваны ошибками перехода, а не трансверсия.
Уход
Предполагается, что после постановки диагноза пациенты будут регулярно наблюдаться кардиологом, эндокринологом, дерматологом и другими специалистами соответствующего профиля по мере появления симптомов.
Людям с синдромом, способным иметь детей, рекомендуется обратиться за генетической консультацией, прежде чем принимать решение о наличии детей. Поскольку синдром часто проявляется как Forme fruste (неполная или необычная форма) необходимо провести обследование всех членов семьи.[13] Поскольку это аутосомно-доминантный признак, вероятность того, что каждый ребенок родится с этим синдромом, составляет пятьдесят процентов. Несмотря на полную пенетрантность, поскольку синдром имеет переменную выраженность, одно поколение может иметь умеренное проявление синдрома, в то время как следующее может быть серьезно затронуто.
После принятия решения о рождении детей и зачатия плода во время беременности проводят наблюдение за состоянием сердца. Если обнаруживается грубая сердечная аномалия, родители получают консультацию о продолжении беременности.
Другое лечение - это плановая помощь при наличии симптомов:[13]
- Для людей с эндокринными проблемами (низкий уровень тиреотопин [гормон гипофиза, отвечающий за регулирование гормонов щитовидной железы], фолликулостимулирующего гормона ) рекомендуется медикаментозная терапия.
- Тем, кого беспокоит появление лентиго, может оказаться полезной криохирургия. Из-за большого количества лентиго это может занять много времени. Может помочь альтернативное лечение кремами с третиноином или гидрохиноном.
- Медикаментозная терапия для людей с сердечными аномалиями, поскольку эти аномалии становятся достаточно серьезными, чтобы оправдать использование этих методов лечения. ЭКГ обязательны перед любым хирургическим вмешательством из-за возможных аритмия.
Прогноз
Сам по себе NSML не является опасным для жизни диагнозом, большинство людей, которым поставлен диагноз, живут нормальной жизнью. Обструктивная кардиомиопатия и другие патологические изменения, затрагивающие сердечно-сосудистую систему, могут быть причиной смерти у тех, у кого сердечная деформация серьезна.[13]
Эпидемиология
В различной литературе этот синдром описывается как «редкий».[13] или «крайне редко».[14] Эпидемиологические данные о том, сколько людей страдают этим синдромом во всем мире, отсутствуют; однако в медицинской литературе описано около 200 случаев.[15]
История
Цейслер и Беккер впервые описали синдром с множественными лентиго, гипертелоризм, pectus carinatum (выступающая грудина) и прогнатизм (протрузия нижней челюсти) в 1936 г.[16] Спорадические описания добавлялись с годами. В 1962 году впервые с этим состоянием были связаны сердечные аномалии и низкий рост.[17] В 1966 году были добавлены три семейных случая: мать, ее сын и дочь.[18] Еще один случай матери двух разных детей с разным отцовством двух детей был добавлен в 1968 году.[19]
Считалось еще в 2002 году.[20] что синдром Нунана с множественными лентигоами (NSML) был связан с нейрофиброматоз I типа (синдром фон Реклингхаузена). Фактически, поскольку оба МКБ9 и МКБ10 отсутствует конкретный диагностический код для NSML, диагностический код для NF1 до сих пор иногда используется в диагностических целях, хотя было показано, что ген не связан с NF1 локус.[21]
Смотрите также
Рекомендации
- ^ Джеймс, Уильям; Бергер, Тимоти; Элстон, Дирк (2005). Болезни кожи Эндрюса: клиническая дерматология (10-е изд.). Сондерс. ISBN 0-7216-2921-0.
- ^ Тидиман В.Е., Рауэн К.А. (июнь 2009 г.). «РАСопатии: синдромы развития нарушения регуляции пути Ras / MAPK». Текущее мнение в области генетики и развития. 19 (3): 230–6. Дои:10.1016 / j.gde.2009.04.001. ЧВК 2743116. PMID 19467855.
- ^ Коппин Б.Д., Темпл И.К. (1997). «Синдром множественного лентиго (синдром Леопарда или прогрессирующий кардиомиопатический лентигиноз)». Журнал медицинской генетики. 34 (7): 582–6. Дои:10.1136 / jmg.34.7.582. ЧВК 1051000. PMID 9222968.
- ^ Tullu MS, Muranjan MN, Kantharia VC, et al. (1 апреля 2000 г.). «Нейрофиброматоз-синдром Нунана или синдром ЛЕОПАРДА? Клиническая дилемма». J Postgrad Med. 46 (2): 98–100. PMID 11013475.
- ^ Горлин Р. Дж., Андерсон Р. К., Бло М. (1969). «Синдром множественных лентигенов». Являюсь. J. Dis. Ребенок. 117 (6): 652–62. Дои:10.1001 / архпеди.1969.02100030654006. PMID 5771505.
- ^ Ворон Д.А., Хэтфилд Х.Х., Калхофф РК (1976). «Синдром множественных лентиго. История болезни и обзор литературы». Являюсь. J. Med. 60 (3): 447–56. Дои:10.1016/0002-9343(76)90764-6. PMID 1258892.
- ^ Ягубян М., Паннетон Дж. М., Линдор Н.М., Конти Э, Саркози А., Пиццути А. (апрель 2004 г.). «Синдром ЛЕОПАРДА: новая ассоциация полианевризмы и обновленная информация о молекулярной генетике заболевания». J. Vasc. Surg. 39 (4): 897–900. Дои:10.1016 / j.jvs.2003.11.030. PMID 15071461.
- ^ Учар С., Калискан Ю., Мартини С., Хейнриц В. (март 2006 г.). «Острый миеломоноцитарный лейкоз у мальчика с синдромом LEOPARD (положительная мутация гена PTPN11)». J. Pediatr. Гематол. Онкол. 28 (3): 123–5. Дои:10.1097 / 01 м / ч.0000199590.21797.0b. PMID 16679933. S2CID 21559684.
- ^ Тарталья М., Мартинелли С., Стелла Л. и др. (2006). «Разнообразие и функциональные последствия мутаций зародышевой линии и соматического PTPN11 при заболеваниях человека». Американский журнал генетики человека. 78 (2): 279–90. Дои:10.1086/499925. ЧВК 1380235. PMID 16358218.
- ^ Hanna N, Montagner A, Lee WH и др. (2006). «Снижение активности фосфатазы SHP-2 при синдроме LEOPARD: последствия связывания PI3K на Gab1». FEBS Lett. 580 (10): 2477–82. Дои:10.1016 / j.febslet.2006.03.088. PMID 16638574. S2CID 27676871.
- ^ Контаридис М.И., Суонсон К.Д., Дэвид Ф.С., Барфорд Д., Нил Б.Г. (2006). «Мутации PTPN11 (Shp2) при синдроме LEOPARD имеют доминантно отрицательные, а не активирующие эффекты». J. Biol. Chem. 281 (10): 6785–92. Дои:10.1074 / jbc.M513068200. PMID 16377799.
- ^ Digilio MC, Sarkozy A, de Zorzi A, et al. (2006). «Синдром ЛЕОПАРДА: клинический диагноз на первом году жизни». Американский журнал медицинской генетики. 140 (7): 740–6. Дои:10.1002 / ajmg.a.31156. PMID 16523510. S2CID 19570040.
- ^ а б c d ЛЕОПАРД-синдром в eMedicine
- ^ «Синдром ЛЕОПАРДА». НОРД - Национальная организация по редким заболеваниям.
- ^ «Синдром Нунана с множественными лентигоами». Национальная медицинская библиотека США.
- ^ Цейслер Е.П., Беккер С.В. (1936). «Генерализованное лентиго: его отношение к системным непрямым невусам». Arch Dermatol Syphilol. 33: 109–125. Дои:10.1001 / archderm.1936.01470070112010.
- ^ Мойнахан EJ (1962). «Множественные симметричные родинки с психическим и соматическим инфантилизмом и гипоплазией гениталий: первый случай нового синдрома у мужчин». Труды Королевского медицинского общества. 55 (11): 959–960. Дои:10.1177/003591576205501112. ЧВК 1896920. PMID 19994192.
- ^ Вальтер Р. Дж., Полански Б. Дж., Гротис И. А. (1966). «Электрокардиографические отклонения в семье с генерализованным лентиго». N. Engl. J. Med. 275 (22): 1220–5. Дои:10.1056 / NEJM196612012752203. PMID 5921856.
- ^ Мэтьюз Н.Л. (1968). «Лентиго и электрокардиографические изменения». N. Engl. J. Med. 278 (14): 780–1. Дои:10.1056 / NEJM196804042781410. PMID 5638719.
- ^ Национальная медицинская библиотека MeSH: C05.660.207.525
- ^ Альбом Б.Е., Даль Н., Зеттерквист П., Аннерен Г. (1995). «Синдром Нунана с пятнами кофе с молоком и синдром множественного лентиго не связаны с локусом нейрофиброматоза 1 типа». Clin. Genet. 48 (2): 85–9. Дои:10.1111 / j.1399-0004.1995.tb04061.x. PMID 7586657. S2CID 31291484.
внешняя ссылка
| Классификация | |
|---|---|
| Внешние ресурсы |