Восстановление карбонила - Carbonyl reduction
В органическая химия, восстановление карбонила это органическое восстановление любой карбонил группа по Восстановитель.
Типичные карбонильные соединения: кетоны, альдегиды, карбоновые кислоты, сложные эфиры, и галогенангидриды. Карбоновые кислоты, сложные эфиры и галогенангидриды могут быть восстановлены либо до альдегидов, либо на следующей стадии до первичные спирты, в зависимости от силы восстановителя; альдегиды и кетоны могут быть восстановлены соответственно до первичных и вторичные спирты. В деоксигенация, алкоголь можно дополнительно уменьшить и полностью удалить.
Гидриды металлов на основе бора и алюминия являются обычными восстановителями; каталитический гидрирование также является важным методом восстановления карбонилов. До открытия растворимых гидридных реагентов, сложные эфиры восстанавливались Редукция Буво – Блана,[1][2][3] с использованием смеси металлического натрия в присутствии спиртов.[4][5]
Производные карбоновых кислот, альдегиды и кетоны до спиртов
Механизм восстановления гидрида
Механизм
В механизм реакции за гидрид металла сокращение основано на нуклеофильное присоединение гидрида к карбонильному углероду. В некоторых случаях катион щелочного металла, особенно Li+, активирует карбонильную группу за счет координации с кислородом карбонила, тем самым усиливая электрофильность карбонила.
Для восстановления производных карбоновой кислоты после восстановления ионом гидрида алюминия удаление приводит к альдегидному продукту (который может быть восстановлен во второй раз до спирта):

Для восстановления альдегидов и кетонов ион гидрида алюминия восстанавливает соединение с образованием соли алкоксида. После полного восстановления алкоксид протонируют с образованием спиртового продукта:

Тенденции реакционной способности карбонила

Кетоны менее реакционноспособны, чем альдегиды, из-за больших стерических эффектов и из-за того, что дополнительная алкильная группа может передавать электронную плотность частичному положительному заряду полярной связи C = O.[6] Следовательно, альдегиды восстанавливаются легче, чем кетоны, и требуют более мягких реагентов и более мягких условий. Карбоновые кислоты и сложные эфиры дополнительно стабилизируются за счет присутствия второго атома кислорода, который может отдавать неподеленную пару в уже полярную связь C = O. Ацилгалогениды наименее стабильны из карбонилов, поскольку галогениды бедны. доноры электронов, а также большой уходящие группы.[7]
Результатом этих тенденций в реакционной способности карбонила является то, что галогенангидриды, кетоны и альдегиды обычно являются наиболее легко восстанавливаемыми соединениями, тогда как кислоты и сложные эфиры требуют более сильного восстановления.
Тенденции реакционной способности гидридов металлов
Четыре основных фактора влияют на эффективность восстановителей на основе гидридов металлов. Во-первых, способность противоиона активировать карбонилы зависит от того, насколько хорошо он может координироваться с кислородом карбонила. Литий меньше и более электрофилен, чем натрий, поэтому он гораздо сильнее координирует свои действия и больше активирует карбонил.[8] Металлы, которые могут иметь несколько зарядов (например, Mg, Al и Zn), образуют катионы с высокой плотностью заряда и, следовательно, также являются более сильными активаторами, чем Na+.[9]
Во-вторых, центральный металл может влиять на прочность восстановителя. Алюминий крупнее бора, поэтому он слабее связывается с гидридами, которые более подвержены атакам; поэтому гидриды алюминия являются лучшими восстановителями, чем боргидриды.[10] Третий фактор, стерильность, делает некоторые замещенные гидриды (гидриды, в которых один или несколько гидридов заменены заместителями) гораздо более слабыми восстановителями, чем гидриды других металлов: триацетоксиборгидрид натрия (NaBH (OAc)3), например, можно использовать для селективного восстановления альдегидов и оставить менее реакционноспособные кетоны непрореагировавшими.[11]
Наконец, заместители могут иметь другие эффекты на реакционную способность восстановителя: ацетоксигруппы препятствуют восстановительной способности NaBH (OAc)3 не только через стерический объем, но и потому, что они улавливают электроны. Группы циано также препятствуют восстановителям, в то время как электронодонорные группы, такие как алкильные группы, могут улучшать их, например, в супергидрид (триэтилборгидрид лития), который является достаточно сильным нуклеофилом, чтобы предотвратить нежелательные перегруппировки во время восстановления.
Из-за этих эффектов заместителей NaBH3CN является очень плохим восстановителем при умеренном pH (> 4), поэтому он предпочитает восстановительное аминирование восстановлению карбонила, как показано ниже:
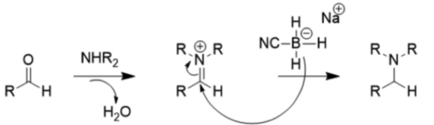
Относительно слабый восстановитель боргидрид натрия обычно используется для восстановления кетонов и альдегидов, потому что в отличие от литийалюминийгидрида он допускает использование многих функциональных групп (нитрогруппа, нитрил, сложный эфир) и может использоваться с водой или этанолом в качестве растворителей.[12] Литийалюминийгидрид и другие сильные восстановители, такие как диизобутилалюминийгидрид, L-селектрид, диборан, диазен и алюмогидрид, также могут восстанавливать альдегиды и кетоны, но не являются предпочтительными, поскольку они опасны и очень реакционноспособны.[13] Однако эти соединения полезны для восстановления карбоновых кислот и сложных эфиров до спиртов, поскольку боргидрид натрия недостаточно мощный для этого.
В следующей таблице показано, какие карбонильные функциональные группы могут быть восстановлены с помощью восстанавливающих агентов (эффективность некоторых из этих реагентов различается в зависимости от условий реакции):

Производные карбоновых кислот до альдегидов
Использование гидридов металлов
Образование альдегидов из производных карбоновых кислот часто является проблемой, поскольку более слабые восстановители (NaBH4) не способны восстанавливать сложные эфиры и карбоновые кислоты, которые относительно стабильны, и более сильные восстановители (LiAlH4) немедленно восстановите образовавшийся альдегид до спирта.[14]
Поскольку хлорангидриды менее стабильны, чем альдегиды и кетоны, они часто используются в сочетании со стерически затрудненными донорами ангидрида при синтезе альдегидов, поскольку относительно слабый восстановитель будет реагировать преимущественно с исходным хлорангидридом, оставляя альдегидный продукт непрореагировавшим. Восстановитель ДИБАЛ-Н (Гидрид диизобутилалюминия) часто используется для этой цели: хотя он обычно восстанавливает все карбонилы, он может перестать восстанавливать альдегид, если используется только один эквивалент при низких температурах.[15] LiAl (OtBu)3 (образован из LiAlH4 и tBuOH in situ) также может останавливать восстановление альдегида посредством механизма, аналогичного DIBAL-H.[16]
Альтернативные методы
Традиционный метод образования альдегидов без восстановления до спиртов - с использованием затрудненных гидридов и реакционноспособных карбонилов - ограничен его узким объемом субстрата и большой зависимостью от условий реакции. Один из способов избежать этого метода - полностью восстановить производное карбоновой кислоты до спирта, а затем снова окислить спирт до альдегида. Другие альтернативы включают формирование тиоэфир или амид Вайнреба с последующим восстановлением новых частиц до альдегида посредством восстановления Фукуямы или реакции Вайнреба, соответственно, или с использованием каталитического гидрирования, как в реакции Розенмунда.

в Снижение Фукуямы карбоновая кислота сначала превращается в тиоэфир путем добавления тиола (с механизмом, аналогичным этерификация ).[17] Затем сложный тиоэфир восстанавливают до альдегида силилгидридом с палладиевым катализатором.
в Реакция Вайнреба, ацилхлорид сначала превращается в амид Вайнреба, затем обрабатывается металлоорганическим реагентом с образованием кетона или алюмогидрида лития с образованием альдегида:[18]

Амид Вайнреба восстанавливается через стабильную хелат, а не электрофильный карбонил, который образуется в результате восстановления гидрида металла; поэтому хелат восстанавливается только один раз, как показано ниже:

В Реакция Розенмунда восстанавливает ацилхлориды до альдегидов с использованием газообразного водорода с катализатором палладий на сульфате бария, небольшая площадь поверхности которого предотвращает чрезмерное восстановление.[19]
Альдегиды и кетоны в алканы

Альдегиды и кетоны восстанавливаются не только до спиртов, но и до алканов. Некоторые реакции для этого преобразования включают Редукция Клемменсена (в сильнокислой среде) и Редукция Вольфа-Кишнера (в строго основных условиях), а также различные модификации реакции Вольфа-Кишнера. В модификации Калиоти, например, используется тозилгидразон с донором гидрида в более мягких условиях без основания;[20] Модификация Майерса заменяет гидразин бис (трет-бутилдиметилсилил) -гидразином, использует более мягкие условия при комнатной температуре, работает быстро и эффективно.[21]
α, β-ненасыщенные карбонилы
В α, β-редукция (также называемый сопряженная редукция) субстрат представляет собой α, β-ненасыщенный карбонил, Enone или же Enal.
При восстановлении этих субстратов происходит 1,2-восстановление, которое дает аллиловый спирт - конкурирует с 1,4-восстановлением, которое образует насыщенный кетон или альдегид. Следующие NaBH4 восстановление енона показывает два возможных продукта: первый из 1,4-восстановления и второй из 1,2-восстановления.[12]

Чем более стерически затруднен еноновый субстрат, тем более вероятно 1,2-восстановление.[12] Кроме того, чтобы избирательно образовывать спирт и избегать продукта 1,4, Луч реакция использует меньшую молекулу Ce (BH4)3 (получено из NaBH4 и CeCl3 объединены in situ) в качестве источника гидрида.[22]
Источник гидрида Zn (BH4)2 также показывает 1,2-селективность, а также большую диастереоселективность; он делает это путем координации не только с кислородом карбонила, но и с соседними атомами:[23]

Стереоселективность
Диастереоселективное сокращение
При восстановлении циклогексанонов источник гидрида может атаковать в осевом направлении для производства экваториального спирта или экваториально для производства аксиального спирта. При осевой атаке (показано красным) гидрид сталкивается с 1,3-диаксиальная деформация. При экваториальной атаке (показано синим) гидрид избегает 1,3-диаксиального взаимодействия, но субстрат подвергается неблагоприятному воздействию. скручивающая деформация когда вновь образованный спирт и добавленный атом водорода затмевают друг друга в промежуточном продукте реакции (как показано в проекции Ньюмана для аксиального спирта).

Большие восстановители, такие как LiBH (Me2ЧЧМе)3, тормозятся 1,3-осевым взаимодействием и поэтому атакуют экваториально.[12] Небольшие восстановители, такие как NaBH4, предпочтительно атакуют в осевом направлении, чтобы избежать затменных взаимодействий, потому что 1,3-диаксиальное взаимодействие для малых молекул минимально; Стереоэлектронные причины также приводились в пользу осевого предпочтения небольших восстановителей.[24] Однако увеличение объема субстрата (и усиление 1,3-осевого взаимодействия) снижает распространенность аксиальных атак даже для небольших доноров гидридов.[25]
Энантиоселективное восстановление
Когда асимметричные кетоны восстанавливаются, образующийся вторичный спирт имеет хиральный центр, которым можно управлять с помощью хиральных катализаторов.
Хорошо известные карбонильные восстановления в асимметричный синтез являются Асимметричное гидрирование Нойори (восстановление бета-кетоэфира / Ru / BINAP) и Сокращение CBS (BH3, хиральный катализатор на основе пролина).
Смотрите также
- пекарские дрожжи, а биотрансформация маршрут для восстановления карбонила.
Рекомендации
- ^ Буво, Луи; Блан, Гюстав Луи (1903). "Подготовка первоочередных спиртов в соответствии с мойеной кислот" [Получение первичных спиртов с помощью соответствующих кислот]. Компт. Ренд. (На французском). 136: 1676–1678.
- ^ Буво, Луи; Блан, Гюстав Луи (1903). «Получение первичных спиртов с соответствующими кислотами» [Получение первичных спиртов с помощью соответствующих кислот]. Компт. Ренд. (На французском). 137: 60–62.
- ^ Буво, Луи; Блан, Гюстав Луи (1904). "Transformation des acides monobasiques saturés dans les alcools primaires Соответствующие" [Превращение насыщенных одноосновных кислот в соответствующие первичные спирты]. Бык. Soc. Чим. Пт. (На французском). 31: 666–672.
- ^ Моффетт, Роберт Брюс (1953). «2- (1-пирролидил) пропанол». Органический синтез. 33: 82. Дои:10.15227 / orgsyn.033.0082.; Коллективный объем, 4, п. 834
- ^ Макмерри, Джон Э. (1973). «Реакция аннелирования изоксазола: 1-метил-4,4a, 5,6,7,8-гексагидронафталин-2 (3ЧАС)-один". Органический синтез. 53: 70. Дои:10.15227 / orgsyn.053.0070.; Коллективный объем, 6, п. 781
- ^ Рош, Алекс. «Кетоны и альдегиды» (PDF). Университет Рутгерса. Получено 1 декабря, 2015.
- ^ Клейден, Джонатан (2012). Органическая химия. ОУП Оксфорд. п. 200. ISBN 978-0199270293.
- ^ Кениг, Буркхард (2009). «Реакции восстановления» (PDF). Современные методы органического синтеза. Institut für Organische Chemie, Университет Регенсбурга. Получено 1 декабря, 2015.
- ^ Кокс, Лиам (2007). «Реакции нуклеофильного присоединения альдегидов и кетонов» (PDF). Бирмингемский университет. Получено 1 декабря, 2015.
- ^ Свитинг, Линда М. (2001). «Восстановители». Тоусонский университет. Архивировано из оригинал 16 ноября 2015 г.. Получено 1 декабря, 2015.
- ^ Гриббл, Гордон В .; Фергюсон, Дункан С. (январь 1975 г.). «Реакции боргидрида натрия в кислой среде. Селективное восстановление альдегидов триацетоксиборгидридом натрия». Журнал химического общества, химические коммуникации. 0 (13): 535–536. Дои:10.1039 / C39750000535.
- ^ а б c d Банфи, Лука; Нарисано, Энрика; Рива, Рената (01.01.2001). Боргидрид натрия. John Wiley & Sons, Ltd. Дои:10.1002 / 047084289x.rs052. ISBN 9780470842898.
- ^ Чайкин, Саул В .; Браун, Велдон Г. (1949-01-01). «Восстановление альдегидов, кетонов и хлоридов кислот боргидридом натрия». Журнал Американского химического общества. 71 (1): 122–125. Дои:10.1021 / ja01169a033. ISSN 0002-7863.
- ^ Гейлорд, Норман Г. (1957-08-01). «Восстановление комплексными гидридами металлов». Журнал химического образования. 34 (8): 367. Bibcode:1957JChEd..34..367G. Дои:10.1021 / ed034p367.
- ^ Захаркин, Л.И .; Хорлина И. М. (1962). «Восстановление сложных эфиров карбоновых кислот до альдегидов гидридом диизобутилалюминия». Буквы Тетраэдра. 3 (14): 619–620. Дои:10.1016 / с0040-4039 (00) 70918-х.
- ^ Кортес, Серджио (2010). «Использование водорода в качестве нуклеофила в восстановлении гидрида» (PDF). Страница д-ра Серхио Кортеса по органической химии. Техасский университет в Далласе. Получено 1 декабря, 2015.
- ^ Фукуяма, Тору; Линь, Шао Чэн; Ли, Лепинг (01.09.1990). «Простое восстановление сложных эфиров этилтиола до альдегидов: применение для полного синтеза (+) - метилового эфира неотрамицина A». Журнал Американского химического общества. 112 (19): 7050–7051. Дои:10.1021 / ja00175a043. ISSN 0002-7863.
- ^ Нахм, Стивен; Вайнреб, Стивен М. (1981). «N-метокси-н-метиламиды как эффективные ацилирующие агенты». Буквы Тетраэдра. 22 (39): 3815–3818. Дои:10.1016 / s0040-4039 (01) 91316-4.
- ^ Мозеттиг, Эрих; Мозинго, Ральф (01.01.2004). Восстановление хлоридов кислот до альдегидов по Розенмунду. John Wiley & Sons, Inc. Дои:10.1002 / 0471264180.or004.07. ISBN 9780471264187.
- ^ Caglioti, L .; Маги, М. (1963-01-01). «Реакция тозилгидразонов с алюмогидридом лития». Тетраэдр. 19 (7): 1127–1131. Дои:10.1016 / S0040-4020 (01) 98571-0.
- ^ Furrow, Michael E .; Майерс, Эндрю Г. (2004-05-01). «Практические процедуры получения N-трет-бутилдиметилсилилгидразонов и их использование в модифицированных восстановлениях Вольфа-Кишнера и в синтезе винилгалогенидов и гем-дигалогенидов». Журнал Американского химического общества. 126 (17): 5436–5445. Дои:10.1021 / ja049694s. ISSN 0002-7863. PMID 15113215.
- ^ Стратегическое применение названных реакций в органическом синтезе (мягкая обложка) Ласло Курти, Барбара Чако ISBN 0-12-429785-4
- ^ Гривс, Ник (2015). «Диастереоселективное снижение кетонов». ChemTube3D. Ливерпульский университет. Получено 1 декабря, 2015.
- ^ Вонг, Стивен С .; Паддон-Роу, Майкл Н. (январь 1990 г.). «Теоретические доказательства в поддержку электронной модели Ань-Эйзенштейна в управлении α-лицевой стереоселективностью в нуклеофильных добавках к карбонильным соединениям». Журнал химического общества, химические коммуникации. 0 (6): 456–458. Дои:10.1039 / c39900000456.
- ^ Krishnamurthy, S .; Браун, Герберт К. (1976-05-01). «Трисиамилборгидрид лития. Новый стерически затрудненный реагент для восстановления циклических кетонов с исключительной стереоселективностью». Журнал Американского химического общества. 98 (11): 3383–3384. Дои:10.1021 / ja00427a061. ISSN 0002-7863.