Открытие и разработка ингибиторов протеазы ВИЧ - Discovery and development of HIV-protease inhibitors
Многие основные физиологические процессы зависят от регуляции протеолитический фермент активности, и могут быть драматические последствия, когда равновесие между ферментом и его субстраты беспокоит. В этой перспективе открытие малых молекул лиганды, подобно ингибиторы протеазы, который может модулировать каталитическую активность, имеет огромный терапевтический эффект.[1] Следовательно, подавление Протеаза ВИЧ является одним из наиболее важных подходов к терапевтическому вмешательству в ВИЧ инфекционное заболевание[2] и их разработка считается главным успехом дизайн лекарств на основе структуры.[3] Они очень эффективны против ВИЧ.[4] и с 1990-х годов были ключевым компонентом антиретровирусной терапии ВИЧ /СПИД.[5]
История
Вирус иммунодефицита человека (ВИЧ) - это лентивирус который имеет два основных вида, ВИЧ-1 что вызывает большинство эпидемия, и ВИЧ-2, близкий родственник, распространение которого сосредоточено в Западной Африке.[6] ВИЧ инфекционное заболевание был впервые описан в 1981 году в Сан-Франциско и Нью-Йорке.[7] В 1985 году ВИЧ был идентифицирован как возбудитель синдрома приобретенного иммунодефицита (СПИД) и его полного геном был доступен сразу. Эти знания открыли путь к развитию селективный ингибиторы.[6]
ВИЧ-2 несет немного меньший риск передачи, чем ВИЧ-1, и инфекция имеет тенденцию медленнее прогрессировать до СПИДа.[7] Обычно термин «ВИЧ» подразумевает ВИЧ-1.[8]
Протеаза ВИЧ-1 - одна из самых известных аспарагиновые протеазы, и привлекательная цель для лечения СПИДа.[9]
После открытия протеазы ВИЧ потребовалось всего 10 лет, чтобы первый ингибитор появился на рынке.[10] Первые отчеты высокоселективной антагонисты против протеазы ВИЧ были обнаружены в 1987 году. Фаза I испытаний саквинавир началось в 1989 году, и это был первый ингибитор протеазы ВИЧ, одобренный для использования по рецепту в 1995 году. Четыре месяца спустя два других ингибитора протеазы, ритонавир и индинавир, были одобрены.[6] В 2009 году десять ингибиторов протеазы вышли на рынок для лечения ВИЧ, кроме одного ингибитора протеазы, ампренавир, был выведен с рынка в 2004 году.[6][11]
Жизненный цикл ВИЧ

ВИЧ принадлежит к классу вирусов, называемых ретровирусы, которые несут генетическую информацию в виде РНК. ВИЧ заражает Т-клетки которые несут CD4 антиген на их поверхности. Когда ВИЧ заражает свою клетку-мишень, он требует слияния вирусной и клеточной мембран.[12] Первым шагом является взаимодействие между белками оболочки вируса (gp120, gp41) и специфическими рецепторами на поверхности клетки-хозяина (например, рецептором CD4) на клетке-мишени. Затем вирус связывается с хемокин корецепторы CXCR4 или же CCR5, что приводит к конформационным изменениям белков оболочки. Это слияние создает поры, через которые вирусный капсид входит в камеру.[13] После проникновения в клетку РНК вируса обратно транскрибируется в ДНК первым вирусно закодированным фермент, то обратная транскриптаза. Вирусная ДНК попадает в ядро где он интегрирован в генетический материал клетки посредством интегрировать, второй фермент, кодируемый вирусом. Активация клетки-хозяина приводит к транскрипция вирусной ДНК в мРНК. Затем мРНК транслируется в вирусные белки, и третий фермент, кодируемый вирусом, а именно протеаза ВИЧ, необходим для расщепления предшественника вирусного полипротеина на отдельные зрелые белки. Вирусная РНК и вирусные белки собираются на поверхности клетки в новые вирионы. Вирионы бутон из клетки и высвобождаются для заражения других клеток. Все инфицированные клетки в конечном итоге погибают из-за этого обширного повреждения клеток, от разрушения генетической системы хозяина до образования почки и высвобождения вирионов.[12]
Механизм действия
В жизненном цикле ВИЧ есть несколько этапов, которым можно помешать, что остановит репликацию вируса. Очень важным этапом является протеолитическое расщепление предшественников полипептидов на зрелые ферменты и структурные белки. катализированный протеазой ВИЧ.[12] Ингибиторы протеазы ВИЧ представляют собой пептидоподобные химические вещества, которые конкурентно ингибируют действие вирусной аспартилпротеазы. Эти препараты предотвращают протеолитическое расщепление полипротеинов Gag и Pol ВИЧ, которые включают важные структурные и ферментативные компоненты вируса. Это предотвращает превращение частиц ВИЧ в их зрелую инфекционную форму.[6]
Ингибиторы протеазы могут изменять адипоцит метаболизм, вызывающий липодистрофия, обычный побочный эффект связаны с использованием большинства ингибиторов протеазы ВИЧ. Было предложено множество механизмов, например, ингибирование адипоцитов. дифференциация, триглицерид накопление и увеличение липолиз. Теории, рассматривающие влияние ингибиторов протеазы на инсулино-стимулированное поглощение глюкозы, также были связаны с липодистрофическим синдромом. Не исключено, что ингибиторы протеаз могут вызвать снижение инсулин -стимулированный тирозин фосфорилирование IRS-1, представляющий ингибирование ранних стадий передачи сигналов инсулина. Уменьшено адипонектин секреция и индуцированная экспрессия интерлейкин-6 связанные с ингибиторами протеазы ВИЧ, также могут способствовать ингибированию инсулино-стимулированного захвата глюкозы.[14]
Дизайн
Ингибиторы протеазы были разработаны для имитации переходное состояние действительной протеазы субстраты. А пептидная связь состоящий из –NH-CO- заменен гидроксиэтиленовой группой (-CH2-CH (OH) -), который протеаза не может расщеплять. Ингибиторы протеазы ВИЧ подходят для активный сайт аспарагиновой протеазы ВИЧ и были рационально разработаны с использованием знаний об аспарагиновой протеазе. образ действий. Наиболее многообещающим имитатором переходного состояния был гидроксиэтиламин, который привел к открытию первого ингибитора протеазы, саквинавир. После этого открытия другие ингибиторы протеазы ВИЧ были разработаны с использованием того же принципа.[15]
Сайт привязки

Протеаза ВИЧ представляет собой C2-симметричный гомодимерный фермент, состоящий из двух 99 аминокислота мономеры. Каждый мономер вносит свой вклад аспарагиновая кислота остаток, необходимый для катализа,[6] Асп-25 и Асп-25´. Протеаза ВИЧ имеет последовательность Asp-Thr -Gly, который является консервативным среди других ферментов аспарагиновой протеазы млекопитающих. Расширенный бета-лист область на мономерах, известная как лоскут, частично составляет сайт связывания субстрата с двумя остатками аспартила, лежащими на дне гидрофобный полость.[12][16][17] Каждый гибкий клапан содержит три характерных участка: боковые цепи, выходящие наружу (Встретились 46, Phe 53), гидрофобные цепи, идущие внутрь (Иль 47, Ile54) и богатая глицином область (Gly48, 49, 51, 52). Ile50 остается на вершине разворота, и когда фермент не связывается, молекула воды делает водородные связи к основной цепи Ile50 на каждом мономере.[17]
Протеазы ВИЧ катализируют гидролиз пептидных связей с высокой селективностью последовательности и каталитическим мастерством. Механизм протеазы ВИЧ имеет много общих черт с остальным семейством аспарагиновых протеаз, хотя полный подробный механизм этого фермента полностью не изучен.[12] Молекула воды, по-видимому, играет роль в открытии и закрытии створок, а также в увеличении сродства между ферментом и субстратом. Аспартильные остатки участвуют в гидролизе пептидных связей.[17] Предпочтительным сайтом расщепления для этого фермента является N-концевой стороне остатков пролина, особенно между фенилаланином и пролином или тирозином, и пролин.[6][16]
Разработка
Первый ингибитор протеазы ВИЧ, саквинавир, представляет собой пептидомиметик гидроксиэтиламин[6] и был продан в 1995 году.[18] Это переходное состояние аналог нативного субстрата протеазы.[6] Наблюдение за тем, что протеаза ВИЧ-1 расщепляет последовательности, содержащие дипептиды Tyr-Pro или Phe-Pro, было основным критерием дизайна.[19] Добавление группы декагидроизохинолина (DIQ) было одной из наиболее значительных модификаций, которые привели к открытию саквинавира. Этот заместитель улучшает растворимость в воде и эффективность за счет ограничения конформационной свободы ингибитора.[20] Саквинавир эффективен как против ВИЧ-1, так и против ВИЧ-2.[5] и обычно хорошо переносится, но высокая концентрация в сыворотке не достигается.[11]
Ритонавир, пептидомиметический ингибитор протеазы ВИЧ, поступил в продажу в 1996 году.[18] Он был разработан, чтобы соответствовать C2-симметрии в сайте связывания протеазы.[6] Разработчики ритонавира, Abbott Laboratories, начали с соединений, которые были активны против вируса, но не имели биодоступность. Были сделаны некоторые улучшения, например, были удалены концевые фенильные остатки и пиридил группы ставят вместо добавления растворимости в воде. Конечным продуктом этих улучшений был ритонавир.[19] Существенные побочные эффекты со стороны желудочно-кишечного тракта и большое количество таблеток являются основными недостатками ритонавира, и поэтому он не используется в качестве единственного средства лечения.[11] Однако это сильный ингибитор метаболизма, опосредованного ферментом цитохрома P450.[19] и он используется только в комбинированной терапии с другими ингибиторами протеазы для фармакокинетического усиления.[11]
Индинавир, который является пептидомиметическим ингибитором протеазы гидроксиэтилена ВИЧ, появился на рынке в 1996 году.[6][18] При разработке индинавира использовалось молекулярное моделирование и рентгеновский снимок Кристальная структура ингибированного ферментного комплекса. Концевые фенильные составляющие способствуют гидрофобному связыванию для увеличения потенция.[19] Это аналог сайта расщепления фенилаланином и пролином полипротеина Gag ВИЧ.[6]
Нелфинавир был первым ингибитором протеазы, который не был пептидомиметиком. В процессе разработки нелфинавира, перорального биодоступного и непептидного ингибитора, был использован повторный анализ сокристаллической структуры белка пептидных ингибиторов, и части ингибиторов были заменены непептидными заместителями.[19] Нелфинавир содержит новую 2-метил-3-гидроксибензамидную группу, тогда как его карбоксильный терминал содержит ту же группу DIQ, что и саквинавир.[19] Нелфинавир поступил в продажу в 1997 году.[18] и был первым ингибитором протеазы, показанным для педиатрический СПИД.[19]
Ампренавир появился на рынке в 1999 году.[18] Это N,N-изамещенный амино-сульфонамид непептидный ингибитор протеазы ВИЧ[6] и имеет некоторые общие черты с предыдущими ингибиторами протеазы. Его ядро аналогично ядру саквинавира, но отличается функциональные группы на обоих концах. На одном конце есть тетрагидрофуран карбаматная группа, а на другом конце - изобутилфенилсульфонамид с добавленным амидом. Эта структура приводит к меньшему количеству хиральный центров, что облегчает синтез и повышает растворимость в водной среде. Это, в свою очередь, обеспечивает лучшую биодоступность при приеме внутрь.[19] Однако ампренавир был снят с продажи в 2004 году, поскольку фосампренавир, его пролекарство, оказался превосходным во многих аспектах.[6]
Лопинавир был продан в 2000 году[18] и изначально был разработан для уменьшения взаимодействия ингибитора с Вал 82 протеазы ВИЧ-1, остаток, который часто мутировавший в лекарственно устойчивый напряжения вируса.[19] Это пептидомиметический ингибитор протеазы ВИЧ.[6] и его ядро идентично таковому ритонавиру. Вместо 5-тиазолил Конечная группа в ритонавире, лопинавир имеет феноксиацетильную группу, а 2-изопропилтиазолильная группа в ритонавире была заменена модифицированным валином, в котором аминоконцевой конец имел шестичленный циклический мочевина прикрепил.[19]
Фосампренавир был продан в 2003 году[18] и представляет собой пролекарство на основе фосфоэфира, которое быстро и широко метаболизируется до ампренавира.[21] Растворимость и биодоступность лучше, чем у ампренавира.[6] что приводит к снижению ежедневного приема таблеток.[22]
Атазанавир был продан в 2003 году[18] и является ингибитором азапептидной протеазы[18] разработан с учетом C2-симметрии сайта связывания фермента.[11] Атазанавир показал лучшие профили устойчивости, чем предыдущие ингибиторы протеазы ВИЧ.[4] Он уникален среди других ингибиторов протеазы, поскольку может быть только поглощен в кислой среде.[11]
Типранавир является непептидным ингибитором протеазы ВИЧ-1[11] и вышла на рынок в 2005 году.[18] В отличие от других ингибиторов протеазы ВИЧ, представленных на рынке, типранавир был разработан на основе непептидного кумарин шаблон и его антипротеазная активность была обнаружена высокопроизводительный скрининг.[23] Этот сульфонамид, содержащий 5,6-дигидро-4-гидрокси-2-пирон, был получен в результате отбора 3-замещенных кумаринов и дигидропиронов.[24] Обладает широкой противовирусной активностью против ВИЧ-1, устойчивого к множественным ингибиторам протеаз.[25]
Дарунавир вышла на рынок в 2006 году[18] и является непептидным аналогом ампренавира с критическим изменением в концевой тетрагидрофурановой (THF) группе. Вместо одной группы ТГФ дарунавир содержит две группы ТГФ, слитые в соединении с образованием бис-ТГФ. часть что делает его более эффективным, чем ампренавир. С этим структурным изменением стереохимия вокруг фрагмента бис-THF вызывает ориентационные изменения, что позволяет продолжать связывание с протеазой, которая выработала устойчивость к ампренавиру.[26]
Все ингибиторы протеазы, одобренные FDA, перечислены ниже.
 | 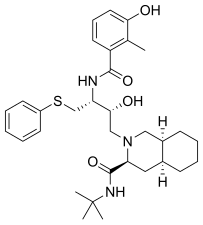 |  |  |
| Саквинавир | Нелфинавир | Ритонавир | Лопинавир |
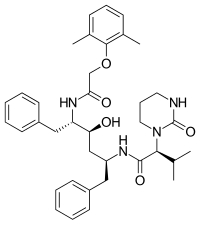
 |  |  | |
| Ампренавир | Фосампренавир | Дарунавир | |
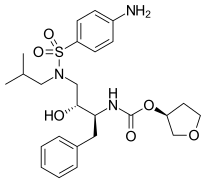
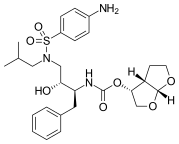
 |  |  | |
| Индинавир | Атазанавир | Типранавир |
Связь структура-деятельность

Все ингибиторы протеазы ВИЧ, представленные на рынке, содержат центральный стержневой мотив, состоящий из гидроксиэтиленового каркаса, за исключением центрального ядра типранавира, который основан на кумариновом каркасе.[15] Очень важная группа ингибиторов протеазы ВИЧ - это гидроксил группа на основном мотиве, которая образует водородную связь с карбоновая кислота на остатки Asp-25 и Asp-25´ в сайте связывания.[16][27] Водородные связи между молекулой воды, которая связана с Ile50 и Ile50 ', и карбонил группы пептидомиметических ингибиторов, по-видимому, связывают их с областями лоскута.[19] С другой стороны, у непептидных ингибиторов есть акцептор протонов, который заменяет четырехкоординированный молекула воды и взаимодействует непосредственно с двумя остатками Ile50 на лоскуте фермента.[28] Специфические карманы в сайте связывания протеазы ВИЧ, часто называемые S1, S1 ', S2 и S2', распознают гидрофобные аминокислоты на природных субстратах. Поэтому эффективность ингибиторов, несущих гидрофобные группы, дополняющие эти области, увеличивается.[29] Некоторые остатки в сайте связывания фермента способны образовывать водородные связи с гидрофильными группами на ингибиторе, например, с фрагментами THF на ампренавире и дарунавире. Поскольку дарунавир имеет фрагмент бис-THF, вместо одного фрагмента THF, как в ампренавире, он может образовывать больше водородных связей и увеличивать энергия связи.[26]
Сопротивление
Мутации, кодирующие изменения конформационной формы, способствуют устойчивости ВИЧ к ингибиторам протеаз.[26] Места этих мутаций в основном находятся в активном сайте фермента протеазы ВИЧ, а также вне активного сайта, в том числе в сайтах расщепления протеазой в предшественниках полипротеина Gag-Pol. Сайты расщепления имеют очень разные последовательности, поэтому протеаза распознает свои субстраты не на основе последовательности, а скорее по консервативной трехмерной форме, которую разделяют субстраты при связывании с активным сайтом. Эта сохранившаяся форма получила название подложка конверт.[30] Было показано, что мутации активного сайта напрямую изменяют взаимодействия ингибиторов и в основном возникают в положениях, где ингибиторы контактируют с остатками протеазы за пределами субстратной оболочки.[31] Считается, что мутации неактивного сайта влияют на другие механизмы, такие как влияние димер стабильность и конформационная гибкость.[32][33]
Более 100 отдельных генов точечные мутации были описаны, из которых по крайней мере 26 специфичны к ингибиторам протеаз. Из них существует около 15 первичных или основных мутаций, которые достаточно значительны, чтобы изменить активность лекарства.[26]В протеазе ВИЧ-1 было обнаружено много мутировавших остатков, которые вызывают лекарственную устойчивость, например, изменение Leu33 на Ile, Val или Phe; Val82 в Ала, Phe, Leu или Thr; Иле84 Валу; и Leu90 в пользу Met.[34] Различные мутации влияют на разные ингибиторы протеаз. Например, мутации в Leu90, очевидно, влияют на саквинавир и нелфинавир, тогда как на активность индинавира влияют мутации в Met46, Val82 и Ile84, а фосампренавир затрагивается, когда Ile50 изменяется на Val и на Ile84. Комбинация мутаций может привести к высокому уровню лекарственной устойчивости, но отдельные мутации обычно не приравниваются к лекарственной устойчивости к ингибиторам протеазы.[26]Мутации можно разделить на первичные и вторичные. Первичные мутации часто лишь незначительно влияют на устойчивость. Химические структуры большинства ингибиторов протеаз очень похожи, поэтому неудивительно, что некоторые первичные мутации одновременно приводят к устойчивости к нескольким ингибиторам протеаз. Перекрестное сопротивление является одной из основных проблем лечения ингибиторами протеазы.[35] Дополнительные мутации, возникающие в протеазе во время непрерывной терапии ингибиторами протеазы, обычно называют вторичными мутациями. Это может привести к высокому уровню устойчивости к ингибиторам протеазы.[35]
Стэнфордская база данных последовательностей ОТ и протеазы ВИЧ (также называемая «База данных по устойчивости к лекарственным препаратам ВИЧ») была сформирована в 1998 году с последовательностями обратной транскриптазы и протеазы ВИЧ от людей с хорошо изученными историями антиретровирусного лечения и является общедоступной для запроса мутаций устойчивости и генотипа. -Лечение, корреляция генотип-фенотип и генотип-результат: http://hivdb.stanford.edu
Хотя субстратная оболочка обеспечивает общую стратегию создания ингибиторов, которые имитируют субстрат и остаются в пределах оболочки, чтобы избежать устойчивости, вызываемой большинством мутаций активного сайта,[36][37] не существует общей стратегии решения проблемы лекарственной устойчивости, особенно из-за тех, кто находится вдали от активного центра. Исследования, направленные на разработку новых методов лечения СПИДа, сосредоточены на предотвращении перекрестной резистентности к лекарствам, которые уже есть на рынке.[12]
Текущее состояние
В январе 2018 года дарунавир был самым последним ингибитором протеазы ВИЧ, появившимся на рынке.[38]
В 2006 г. GlaxoSmithKline прекратил клиническую разработку фазы II бреканавир, исследуемый ингибитор протеазы для лечения ВИЧ, из-за непреодолимых проблем, связанных с составом.[39]
Летом 2009 года GlaxoSmithKline и Concert Pharmaceuticals объявили о своем сотрудничестве по разработке и коммерциализации дейтерий -содержащие лекарства. Одним из них является CTP-518, ингибитор протеазы для лечения ВИЧ, который, как ожидается, войдет в фазу I клинических испытаний во второй половине 2009 года. CTP-518 - новый ингибитор протеазы ВИЧ, разработанный путем замены определенных ключевых атомов водорода атазанавира на дейтерий. Доклинические исследования показали, что эта модификация полностью сохраняет противовирусную активность, но, очевидно, может замедлить метаболизм в печени и тем самым увеличить период полужизни и плазменный уровни желоба. Таким образом, CTP-518 потенциально может стать первым ингибитором протеазы ВИЧ, который устраняет необходимость в одновременном приеме с бустирующим агентом, таким как ритонавир.[40]
Смотрите также
- Антиретровирусный препарат
- Ингибитор обратной транскриптазы
- Ингибитор интегразы
- Ингибитор входа
- Открытие и разработка ненуклеозидных ингибиторов обратной транскриптазы
Рекомендации
- ^ Cuccioloni, M; Mozzicafreddo, M; Bonfili, L; Чекарини, V; Eleuteri, A.M .; Анджелетти, М (2009). «Полифенолы природного происхождения как образец для разработки лекарств. Сосредоточение внимания на сериновых протеазах». Химическая биология и дизайн лекарств. 74 (1): 1–15. Дои:10.1111 / j.1747-0285.2009.00836.x. PMID 19519739.
- ^ Чен, X; Кемпф, Д. Дж .; Ли, Л; Sham, H.L .; Васаванонда, S; Wideburg, N.E .; Салдивар, А; Марш, К. С .; Макдональд, Э; Норбек, Д. В. (2003). «Синтез и исследования SAR мощных ингибиторов протеазы ВИЧ, содержащих новые диметилфеноксилацетаты в качестве лигандов P2». Письма по биоорганической и медицинской химии. 13 (21): 3657–60. Дои:10.1016 / j.bmcl.2003.08.043. PMID 14552751.
- ^ Адачи, М. Охара, Т; Курихара, К; Тамада, Т; Honjo, E; Окадзаки, N; Arai, S; Шояма, Й; Кимура, К; Мацумура, Н; Сугияма, S; Адачи, H; Такано, К; Мори, Y; Хидака, К; Кимура, Т; Хаяси, Й; Кисо, Y; Куроки, Р. (2009). «Структура протеазы ВИЧ-1 в комплексе с сильнодействующим ингибитором KNI-272, определенная методами рентгеновской и нейтронной кристаллографии высокого разрешения». Труды Национальной академии наук. 106 (12): 4641–6. Дои:10.1073 / pnas.0809400106. ЧВК 2660780. PMID 19273847.
- ^ а б Янчунас-младший, J; Langley, D. R .; Тао, L; Rose, R.E .; Фрибург, Дж; Colonno, R.J .; Дойл, М. Л. (2005). «Молекулярная основа повышенной чувствительности изолятов с устойчивостью к атазанавиру, приводящей к замене I50L на другие ингибиторы протеазы». Противомикробные препараты и химиотерапия. 49 (9): 3825–32. Дои:10.1128 / AAC.49.9.3825-3832.2005. ЧВК 1195399. PMID 16127059.
- ^ а б Brower, E.T .; Bacha, U. M .; Кавасаки, Y; Фрейре, Э (2008). «Ингибирование протеазы ВИЧ-2 ингибиторами протеазы ВИЧ-1 при клиническом использовании». Химическая биология и дизайн лекарств. 71 (4): 298–305. Дои:10.1111 / j.1747-0285.2008.00647.x. PMID 18312292.
- ^ а б c d е ж грамм час я j k л м п о п Brunton, L.L .; Lazo, J.S .; Паркер, К. (2006). Гудман и Гилманс: Фармакологические основы терапии (11-е изд.). Макгроу-Хилл.[страница нужна ]
- ^ а б Болезнь ВИЧ в eMedicine
- ^ Куруп, Алка; Мекапати, Суреш; Гарг, Раджни; Хэнш, Корвин (2003). «Ингибиторы протеазы ВИЧ-1: сравнительный анализ QSAR». Современная лекарственная химия. 10 (17): 1679–88. Дои:10.2174/0929867033457070. PMID 12871116.
- ^ Ши, Хайбин; Лю, Кай; Леонг, Венди W.Y .; Яо, Шао К. (2009). «Целесообразный твердофазный синтез симметричных и асимметричных библиотек диолов, нацеленных на аспарагиновые протеазы». Письма по биоорганической и медицинской химии. 19 (14): 3945–8. Дои:10.1016 / j.bmcl.2009.03.041. PMID 19328682.
- ^ Турок, Борис (2006). «Таргетинг на протеазы: успехи, неудачи и перспективы на будущее». Обзоры природы Drug Discovery. 5 (9): 785–99. Дои:10.1038 / nrd2092. PMID 16955069.
- ^ а б c d е ж грамм Грациани, Эми Л. (17 июня 2014 г.). «Ингибиторы протеазы ВИЧ». Своевременно.
- ^ а б c d е ж Брик, А., Вонг, Ч. (2003) протеаза ВИЧ-1: механизм и открытие лекарств. Органическая и биомолекулярная химия. 1(1); 5–14.
- ^ Варнке Д., Баррето Дж. И Темесген З. (2007) Антиретровирусные препараты. Журнал клинической фармакологии. 47(12); 1570–1579.
- ^ Ким, Р.Дж., Уилсон, К.Г., Вабич, М., Лазар, М.А., и Степпан, К.М. (2006) Специфичные для ингибитора протеазы ВИЧ изменения в дифференцировке и метаболизме адипоцитов человека. Ожирение. 14; 994–1002.
- ^ а б Де Клерк, Э. (2009) История антиретровирусных препаратов: ключевые открытия за последние 25 лет. Обзоры в медицинской вирусологии. 19; 287–299.
- ^ а б c Мимото Т., Хаттори Н., Такаку Х. и др. (2000) Взаимосвязь между структурой и активностью пероральных эффективных ингибиторов протеазы ВИЧ на основе трипептида, содержащих гидроксиметилкарбонилизостер. Химико-фармацевтический бюллетень. 48(9); 1310–1326.
- ^ а б c Перес, М.А.С., Фернандес, П.А. и Ramos, M.J. (2007) Дизайн лекарств: новые ингибиторы протеазы ВИЧ-1 на основе нелфинавира в качестве свинца. Журнал молекулярной графики и моделирования. 26; 634–642.
- ^ а б c d е ж грамм час я j k Флекснер, К. (2007) Разработка лекарств от ВИЧ: следующие 25 лет. Обзоры природы Drug Discovery. 6; 959–966.
- ^ а б c d е ж грамм час я j k Влодавер, А. (2002) Рациональный подход к разработке лекарств от СПИДа с помощью структурной биологии. Ежегодный обзор медицины. 53; 595–614.
- ^ Смит, Х.Дж. и Саймонс, К. (2005) Ферменты и их ингибирование: разработка лекарств (6-е издание). Соединенные Штаты Америки: CRC press
- ^ Чепмен, Т.М., Плоскер, Г.Л., Перри, К.М. (2004) Фосампренавир - Обзор его использования в ведении пациентов с ВИЧ-инфекцией, не получавших антиретровирусную терапию. Наркотики. 64; 2101–2124.
- ^ а б c d е Маккой, К. (2007) Дарунавир: непептидный антиретровирусный ингибитор протеазы. Клиническая терапия. 29(8); 1559–1576.
- ^ Лю Ф., Ковалевский А.Ю., Ти Ю., Гош А.К., Харрисон Р.В. и Вебер И.Т. (2008) Влияние мутаций лоскута на структуру протеазы ВИЧ и ингибирование саквинавиром и дарунавиром. Журнал молекулярной биологии. 381(1); 102–115
- ^ Лебон, Ф. и Ледек, М. (2000) Подходы к разработке эффективных ингибиторов протеазы ВИЧ-1. Современная лекарственная химия. 7; 455–477.
- ^ Blum, A. et al. (2008) Ахиральные олигоамины как универсальный инструмент для разработки ингибиторов аспарагиновой протеазы. Биоорганическая и медицинская химия. 16; 8574–8586.
- ^ Prabu-Jeyabalan, Nalivaika E, Schiffer CA. (2002) Форма субстрата определяет специфичность распознавания протеазы ВИЧ-1: анализ кристаллических структур шести субстратных комплексов. «Структура» 10 (3): 369-81.
- ^ King NM, Prabu-Jeyabalan M, Nalivaika EA, Schiffer CA (2004) Борьба с восприимчивостью к лекарственной устойчивости: уроки протеазы ВИЧ-1. Chem Biol. Октябрь; 11 (10): 1333-8.
- ^ Бихани, С. К., Дас, А., Прашар, В., Феррер, Ж.-Л. и Хосур; М.В. (2009) Механизм устойчивости выявлен с помощью кристаллических структур нелигандованных мутантов N88D и N88S с неактивным сайтом протеазы ВИЧ-1, устойчивых к нелфинавиру. Сообщения о биохимических и биофизических исследованиях. 389; 295–300.
- ^ де Вера И.М., Смит А.Н., Танцель М.С., Хуанг Х, Данн Б.М., Фануччи Г.Э. (2013) Биохимия. Выяснение связи между конформационным отбором образцов и лекарственной устойчивостью протеазы ВИЧ-1. 14; 52 (19): 3278-88. DOI: 10,1021 / bi400109d. Epub 2013 1 мая.
- ^ Лемке, Т.Л., Уильямс, Д.А., Рош, В.Ф. и Зито, С. (2008) Принципы медицинской химии Фуа (6-е издание). Соединенные Штаты Америки: Lippincott williams & Wilkins, предприятие Wolters Kluwer.
- ^ а б Маарсевен, Н.В. и Баучер, К. (2008) Устойчивость к антиретровирусным препаратам в клинической практике. Лондон: Mediscript Ltd.
- ^ Kairys V, Gilson MK, Lather V, Schiffer CA, Fernandes MX. (2009) На пути к разработке ингибиторов ферментов, устойчивых к мутациям: дальнейшая оценка гипотезы субстратной оболочки. Chem Biol Drug Des. Сен; 74 (3): 234-45. DOI: 10.1111 / j.1747-0285.2009.00851.x
- ^ Налам М.Н., Али А., Альтман М.Д., Редди Г.С., Челлаппан С., Кайрис В., Озен А., Цао Х., Гилсон М.К., Тидор Б., Рана Т.М., Шиффер К.А. (2010) Оценка гипотезы субстрат-оболочка: структурный анализ новых ингибиторов протеазы ВИЧ-1, разработанных для обеспечения устойчивости к лекарствам. J Virol. 2010 Май; 84 (10): 5368-78. DOI: 10.1128 / JVI.02531-09. Epub 2010 17 марта.
- ^ Де Клерк, Э. (2009) Препараты против ВИЧ: 25 соединений, одобренных в течение 25 лет после открытия ВИЧ. Международный журнал противомикробных агентов. 33; 307–320.
- ^ «Архивная копия». Архивировано из оригинал на 2008-12-03. Получено 2008-06-11.CS1 maint: заархивированная копия как заголовок (связь) GlaxoSmithKline прекращает клиническую разработку исследуемого ингибитора протеазы бреканавира (640385). Проверено 4 ноября. 2009 г.
- ^ «Архивная копия». Архивировано из оригинал 31 августа 2009 г.. Получено 2009-11-05.CS1 maint: заархивированная копия как заголовок (связь)GSK и Concert Pharmaceuticals создают альянс для разработки новых лекарств, модифицированных дейтерием. Проверено 4 ноября. 2009 г.