Синдром Кокейна - Cockayne syndrome
| Синдром Кокейна | |
|---|---|
| Другие имена | Синдром Нейла-Дингуолла |
| Специальность | Медицинская генетика, неврология, дерматология |
Синдром Кокейна (CS), также называемый Синдром Нейла-Дингуолла, является редким и смертельным аутосомный рецессивный нейродегенеративный расстройство, характеризующееся задержкой роста, нарушением развития нервная система, аномальная чувствительность к Солнечный свет (светочувствительность ), глазные заболевания и преждевременное старение.[1][2][3] Неспособность процветать и неврологические расстройства являются критериями для диагностики, тогда как светочувствительность, потеря слуха, глазные аномалии и кариес - другие очень общие признаки.[3] Возможны проблемы с некоторыми или всеми внутренними органами. Это связано с группой расстройств, называемых лейкодистрофии, которые представляют собой состояния, характеризующиеся деградацией неврологических белое вещество. Основное заболевание - это дефект Ремонт ДНК механизм.[4] В отличие от других дефектов репарации ДНК, пациенты с CS не предрасположены к раку или инфекции.[5] Синдром Кокейна - редкое, но разрушительное заболевание, обычно приводящее к смерти в течение первого или второго десятилетия жизни. Мутации определенных генов при синдроме Кокейна известны, но широко распространенные эффекты и их связь с репарацией ДНК еще предстоит хорошо понять.[5]
Он назван в честь английского врача. Эдвард Альфред Кокейн (1880–1956), которые впервые описали его в 1936 году и повторно описали в 1946 году.[6] Синдром Нейла-Дингуолла был назван в честь Мэри М. Дингуолл и Кэтрин А. Нил.[6] Эти два ученых описали случай двух братьев с синдромом Кокейна и заявили, что это была та же болезнь, которую описал Кокейн. В своей статье эти двое способствовали появлению признаков болезни, обнаружив кальцификаты в головном мозге. Они также сравнили синдром Кокейна с тем, что сейчас известно как Синдром прогерии Хатчинсона-Гилфорда (HGPS), тогда называемый прогерией, из-за позднего старения, характерного для обоих заболеваний.[6]
Типы
- CS Тип I, «классическая» форма, характеризуется нормальным ростом плода с появлением аномалий в первые два года жизни. Зрение и слух постепенно ухудшаются.[7] Центральная и периферическая нервные системы постепенно деградируют вплоть до смерти в первом или втором десятилетии жизни в результате серьезной неврологической деградации. Кортикальная атрофия менее выражена при CS I типа.[8]
- CS Тип II присутствует с рождения (врожденный ) и является гораздо более серьезным, чем CS Type 1.[7] После рождения это связано с очень незначительным неврологическим развитием. Смерть обычно наступает к семи годам. Этот специфический тип также был обозначен как церебро-окуло-фациально-скелетный (COFS) синдром или синдром Пена-Шокейра типа II.[7] Синдром COFS назван так из-за воздействия, которое он оказывает на мозг, глаза, лицо и скелетную систему, поскольку заболевание часто вызывает атрофию мозга, катаракту, потерю жира на лице и остеопороз. Синдром COFS можно подразделить на несколько состояний (типы COFS 1, 2, 3 (связанные с пигментная ксеродермия ) и 4).[9] Обычно у пациентов с этой ранней формой заболевания наблюдается более серьезное повреждение головного мозга, включая уменьшение миелинизации белого вещества и более распространенные кальцификаты, в том числе в коре и базальных ганглиях.[8]
- CS Тип III, характеризующийся поздним началом, обычно мягче, чем Типы I и II.[7] Часто пациенты с типом III доживают до взрослого возраста.
- Синдром Xeroderma pigmentosum-Cockayne (XP-CS) возникает, когда человек также страдает пигментной ксеродермией, другим заболеванием восстановления ДНК. Выражены некоторые симптомы каждого заболевания. Например, присутствуют характерные для XP отклонения в виде веснушек и пигментации. Выявлены характерные для CS неврологическое расстройство, спастичность и недоразвитие половых органов. Однако гипомиелинизация и черты лица типичных пациентов с КС отсутствуют.[10]
Причины
Если гипероксия или избыток кислород происходит в нашем теле, наши клеточный метаболизм производят несколько высокореактивных форм кислорода, называемых свободные радикалы. Это может вызвать окислительное повреждение клеточных компонентов, включая ДНК. В нормальных клетках наш организм восстанавливает поврежденные участки. В случае этого заболевания из-за тонких дефектов в транскрипция, детская генетический оборудование для синтеза белки необходимые организму не работают в нормальном режиме. То есть ученые полагали, что генетический механизм этих детей для синтеза белков, необходимых организму, не работает в нормальном режиме. Со временем эта теория пошла на пользу. развивающий неудача и смерть. Каждую минуту организм перекачивает от 10 до 20 литров кислорода через кровь, разнося его в миллиарды клеток нашего тела. В нормальном молекулярный форме кислород безвреден. Однако сотовая метаболизм с участием кислорода может образовываться несколько высокоактивных свободных радикалов. Эти свободные радикалы могут вызывать окислительное повреждение к клеточным компонентам, включая ДНК. В среднем человеческая клетка, несколько тысяч поражения происходят в ДНК каждый день. Многие из этих поражений возникают в результате окислительное повреждение. Каждое поражение - поврежденный участок ДНК - необходимо вырезать, а ДНК восстанавливать, чтобы сохранить ее нормальную функцию. Необработанная ДНК может потерять способность кодировать белки. Также могут возникнуть мутации. Эти мутации могут активировать онкогены или заглушить гены-супрессоры опухоли. Согласно исследованиям, окислительное повреждение активных генов преимущественно не восстанавливается, а в самых тяжелых случаях восстановление замедляется на протяжении всего периода. геном. В результате накопление окислительного повреждения может нарушить нормальные функции ДНК и даже может привести к запуску программы гибели клеток (апоптоза). У детей с этим заболеванием не восстанавливаются активные гены, вызывающие окислительное повреждение. Обычно восстановление после окислительного повреждения происходит быстрее в активных генах (которые составляют менее пяти процентов генома), чем в неактивных областях ДНК. В результате накопление окислительного повреждения может нарушить нормальные функции ДНК и даже может привести к запуску программы гибели клеток (апоптоз ).[нужна цитата ]
Генетика
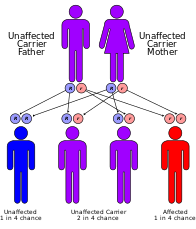
Синдром Кокейна классифицируется генетически следующее:
| Тип | OMIM | Ген |
|---|---|---|
| А | 216400 | ERCC8 (также называется CSA) |
| B | 133540 | ERCC6 (также называется CSB) |
| C | 216411 | никто не известен |
- Мутации в генах ERCC8 (также известных как CSA) или ERCC6 (также известных как CSB) являются причиной синдрома Кокейна.[7] Мутации в гене ERCC6 составляют ~ 70% случаев. Белки, производимые этими генами, участвуют в восстановлении поврежденной ДНК через связанный с транскрипцией механизм репарации, особенно ДНК в активных генах. Повреждение ДНК вызывается ультрафиолетовыми лучами солнечного света, радиацией или свободными радикалами в организме. Нормальная клетка может восстанавливать повреждения ДНК до того, как они накопятся. Если ген ERCC6 или ERCC8 изменен (как при синдроме Кокейна), повреждение ДНК, обнаруженное во время транскрипции, не восстанавливается, в результате чего РНК-полимераза останавливается в этом месте, препятствуя экспрессии гена. По мере накопления невосстановленных повреждений ДНК все более активная экспрессия генов затрудняется, что приводит к неисправным клеткам или их гибели, что, вероятно, способствует появлению таких признаков синдрома Кокейна, как преждевременное старение и гипомиелинизация нейронов.[7]
Механизм
В отличие от клеток с нормальной способностью к восстановлению, CSA и CSB дефицитные клетки не могут предпочтительно восстанавливать циклобутан димеры пиримидина индуцируется действием ультрафиолетового (УФ) света на матричную нить активно записано гены.[11] Этот дефицит отражает потерю способности выполнять процесс восстановления ДНК, известный как связанный с транскрипцией. эксцизионная репарация нуклеотидов (TC-NER).[нужна цитата ]
Внутри поврежденной клетки белок CSA обычно локализуется в участках Повреждение ДНК, особенно межцепочечные сшивки, двухцепочечные разрывы и некоторые моноаддукты.[12] Белок CSB также обычно рекрутируется на участки повреждения ДНК, и его рекрутирование происходит наиболее быстро и надежно, а именно: межцепочечные сшивки> двухцепочечные разрывы> моноаддукты> окислительное повреждение.[12] Белок CSB образует комплекс с другим белком репарации ДНК, SNM1A (DCLRE1A ), а 5 '- 3' экзонуклеаза, который локализуется в межцепочечных перекрестных связях транскрипционно-зависимым образом.[13] Накопление белка CSB на участках двухцепочечных разрывов ДНК происходит транскрипционно-зависимым образом и способствует гомологичный рекомбинационный ремонт поломок.[14] Вовремя G0 /Фаза G1 клеточного цикла, повреждение ДНК может запускать CSB-зависимый процесс рекомбинационной репарации, который использует РНК (скорее, чем ДНК ) шаблон.[15]
Признаки преждевременного старения CS, вероятно, связаны, по крайней мере частично, с недостатками в Ремонт ДНК (видеть Теория повреждений ДНК старения ).[нужна цитата ]
Диагностика
У людей с этим синдромом размер головы меньше нормального (микроцефалия ), имеют небольшой рост (карликовость ), их глаза выглядят запавшими, и они выглядят «постаревшими». У них часто длинные конечности с совместные контрактуры (невозможность расслабить мышцу в суставе), сгорбленная спина (кифоз ), и они могут быть очень тонкими (кахетический ) из-за потери подкожного жира. Их маленький подбородок, большие уши и острый тонкий нос часто придают им пожилой вид.[8]Кожа людей с синдромом Кокейна также часто поражается: гиперпигментация, варикозное расширение вен или сосудистые звездочки (телеангиэктазия ),[8] и серьезная чувствительность к солнечному свету - обычное явление, даже у людей без XP-CS. Часто пациенты с синдромом Кокейна сильно обжигаются или покрываются волдырями при очень небольшом тепловом воздействии. Глаза пациентов могут быть поражены различными способами, и аномалии глаз часто встречаются при CS. Катаракта и помутнение роговицы (помутнение роговицы ) общие. Может произойти потеря и повреждение нервов зрительного нерва, что приведет к атрофии зрительного нерва.[3] Нистагм, или непроизвольное движение глаз, а зрачки, которые не расширяются, демонстрируют потерю контроля над произвольными и непроизвольными движениями мышц.[8] Пигментация сетчатки из соли и перца также является типичным признаком. Диагноз устанавливается с помощью специального теста на восстановление ДНК, который измеряет восстановление РНК после воздействия УФ-излучения. Несмотря на то, что он связан с генами, участвующими в эксцизионная репарация нуклеотидов (NER), в отличие от пигментная ксеродермия, CS не связан с повышенным риском рака.[5]
Лабораторные исследования
У пациентов с синдромом Кокейна облученные УФ-излучением клетки показывают снижение синтеза ДНК и РНК.https://emedicine.medscape.com/article/1115866-workup#c5 Лабораторные исследования в основном полезны для устранения других нарушений. Например, рентгенография скелета, эндокринологические тесты и исследования хромосомных повреждений могут помочь исключить нарушения, включенные в дифференциальный диагноз.[нужна цитата ]
Визуальные исследования
КТ головного мозга у пациентов с синдромом Кокейна может выявить кальцификаты и корковую атрофию.[нужна цитата ]
Прочие тесты
Возможно дородовое обследование. Культивирование клеток амниотической жидкости используется для демонстрации того, что фетальные клетки испытывают дефицит синтеза РНК после УФ-облучения.
Неврология
Визуализирующие исследования показывают широко распространенное отсутствие миелиновых оболочек нейронов в белом веществе мозга и общую атрофию коры.[5] Кальцификации также были обнаружены в скорлупа, площадь передний мозг который регулирует движения и помогает в некоторых формах обучения,[8] вместе с корой.[6] Дополнительно атрофия центральной области мозжечок обнаруженный у пациентов с синдромом Кокейна, также может приводить к отсутствию мышечного контроля, особенно непроизвольному, и обычно наблюдается плохая осанка.[нужна цитата ]
Уход
Постоянного излечения от этого синдрома не существует, хотя пациентов можно лечить симптоматически. Лечение обычно включает физиотерапию и небольшие операции на пораженных органах, например удаление катаракты.[3] Также рекомендуется носить высокоэффективный солнцезащитный крем и защитную одежду, поскольку пациенты с синдромом Кокейна очень чувствительны к УФ-излучению.[16] Также может помочь оптимальное питание. Родителям рекомендуется генетическое консультирование, так как вероятность передачи заболевания любым будущим детям составляет 25%. Также возможно пренатальное тестирование.[3] Другой важный аспект - это предотвращение рецидива CS у других братьев и сестер. Выявление вовлеченных генных дефектов позволяет предлагать генетическое консультирование и дородовое диагностическое тестирование родителям, у которых уже есть один больной ребенок.[17]
Прогноз
В прогноз для людей с синдромом Кокейна это плохо, так как смерть обычно наступает к 12 годам. [18]Прогноз синдрома Кокейна зависит от типа заболевания. В зависимости от тяжести и начала симптомов существует три типа синдрома Кокейна. Однако различия между типами не всегда очевидны, и некоторые исследователи полагают, что признаки и симптомы отражают спектр, а не отдельные типы: синдром Кокейна типа A (CSA) характеризуется нормальным развитием, пока ребенку не исполнится 1 или 2 года. старые, в этот момент рост замедляется и отмечаются задержки в развитии. Симптомы не проявляются до 1 года. Ожидаемая продолжительность жизни для типа А составляет примерно от 10 до 20 лет. Эти симптомы наблюдаются у детей типа 1 CS. Синдром Кокейна типа B (CSB), также известный как «церебро-окулофасцио-скелетный (COFS) синдром» (или «синдром Пена-Шокейра типа B»), является наиболее тяжелым. подтип. Симптомы присутствуют при рождении, и нормальное развитие мозга прекращается после рождения. Средняя продолжительность жизни детей с типом B - до 7 лет. Эти симптомы наблюдаются у детей типа 2 типа CS. Синдром Кокейна типа C (CSC) появляется позже в детстве с более легкими симптомами, чем другие типы, и более медленным прогрессированием расстройства. Люди с этим типом синдрома Кокейна доживают до взрослого возраста, средняя продолжительность жизни составляет от 40 до 50 лет. Эти симптомы наблюдаются при CS 3 типа.[нужна цитата ]
Эпидемиология
Синдром Кокейна встречается во всем мире редко. Не сообщается о расовой предрасположенности к синдрому Кокейна. Для синдрома Кокейна не описано сексуальных предпочтений; соотношение мужчин и женщин одинаковое. Синдром Кокейна I (CS-A) проявляется в детстве. Синдром Кокейна II (CS-B) проявляется при рождении или в младенчестве и имеет худший прогноз.[нужна цитата ]
Недавнее исследование
В недавнем исследовании, проведенном в январе 2018 года, упоминаются различные особенности CS, которые наблюдаются во всем мире, имеют сходства и различия: CS имеет частоту 1 случай на 250 000 живорождений и распространенность 2,5 на миллион, что в значительной степени согласуется в разных регионах мира:[19]
| Затронутые части | Клинические признаки | патология |
|---|---|---|
| Лицо | Морщинистые лица. Запавшие глаза, большие уши, тонкий заостренный нос. Маленький подбородок. Кариес, гипоплазия эмали | |
| Кожа, волосы, гвозди | Светочувствительность. Морщинистая и возрастная кожа. Тонкие сухие волосы, преждевременная седина. Бедные венозный доступ. | |
| Центральная нервная система | Микроцефалия обычно начиная с 2 лет. Умственная отсталость с низкий IQ. Отложенные вехи.Тремор, атаксия, припадки, удары, и субдуральные кровоизлияния. | Демиелинизация - неоднородно и сегментарно - »Метахроматическая лейкодистрофия ". Обе олигодендроглия и Шванновские клетки под действием. Влияет мозговой белое вещество, мозолистое тело, мозговой ствол, спинной мозг, и периферические нервы. Нейронный потеря на нескольких сайтах, особенно мозжечок. Утрата клетки переднего рога из-за антероградного и / или ретроградного вырождение. Кальцификация [55–95%] кора головного мозга (особенно глубины борозды, базальный ганглий, мозжечок, таламус; также из артерии, артериолы, и капилляры. Сосудистый изменения - Струнные сосуды, особенно в областях метахроматической лейкодистрофии, кальцификации в лептоменингеальный сосуды ускоренный атеросклероз и артериолосклероз.Глиоз настоящее. Астроциты и микроглия может показывать нерегулярные цитоплазма, несколько ядра. Может быть замечено как белое вещество высокой интенсивности на FLAIR MRI Последовательность сигналов. Нет большого мозга пороки развития. Наблюдается относительная щадимость коры головного мозга, небольшое истончение корковой ленты. Нормальный круговорот узор с уширением борозды. Ламинирование, размер нейронов и конфигурация неокортекс сохраняются. Может показать теменный затылочный доминирование. тяжелый мозжечок атрофия. Утрата Пуркинье, гранулированный нейроны, а в некоторых случаях нейроны в зубчатое ядро. Дендриты из Клетки Пуркинье могут быть сильно деформированы («цветки кактуса») ожелезненными дендритами. У дендритов меньше ветвей высшего порядка. Пуркинье »аксональный торпеды »Может присутствовать.Желудочковый увеличение, увеличенное цистерна magna. Амилоидные бляшки, нейрофибриллярные сплетения, Тела Хирано не часто встречается, хотя убиквитин реактивность из аксоны настоящее время |
| Слух и вестибулярная система | Нейросенсорная, высокий тон потеря слуха [60–90%]. Смешанные токопроводящие и нейросенсорная тугоухость (44%) Чаще двусторонний, редко односторонний | Потеря волосковых клеток в улитка, особенно в базальный поворот. Потеря нейронов в спиральный узел. Атрофия слуховые пути. Scala communis, утолщенный стремени кураре, расширенный prototympanum. Потеря волосковых клеток в верхней части. Потеря нейронов в вестибулярный ганглий. Крах эндолимфатический проток нижних частей |
| Зрение | Помутнение роговицы. Катаракты [36–86%]. Обычно двусторонний, чаще всего развивается к 4 годам.Пигментная ретинопатия («Соль и перец») [43–89%]. Миотик зрачки, Оптический диск бледность, Энофтальм, Узкий глазные щели. | Периодическая потеря меланин пигмент гранулы. Липофусцин отложение, большие клетки с пигментом в периваскулярный распределение. Сетчатка пигмент эпителиальный атрофия и гиперплазия. Потеря клеток в ганглий и внешние слои ядерных клеток. И внешний, и внутренний сегменты фоторецепторы под действием. Зрительный нерв атрофия, с частичной демиелинизация, потеря аксонов и глиоз |
| Костно-мышечной системы | Кахектик карликовость. Контракты. Кифоз, сколиоз. Сгорбленная поза. Атрофия мышц. | Денервация миопатия, неиспользованная атрофия |
| Сердечно-сосудистая система | Ускоренный гипертония. Дилатация корня аорты. Кардиомиопатия. | Повысился интима медиальное утолщение. Атеросклероз, атеросклероз. |
| Желудочно-кишечная система | Суровый рефлюкс. Аномальный перистальтика желудочно-кишечного тракта. Многие имеют чрескожный гастростомические трубки. Гепатомегалия, спленомегалия, повышенный ферменты печени. Изменено метаболизм наркотиков | - |
| Почечная система | Почечная недостаточность | Почечные артерии показать изменения при запущенном атеросклерозе и артериолосклерозе. Односторонний или гипоплазия почек. |
| Репродуктивная система | - | - |
| Самцы | Микропенис, меньше яичко размер | - |
| Самки | Яичников атрофия. Успешный беременность было сообщено. | - |
| Эндокринные системы | Нормальный вторичный половые признаки. Нормальный гормон роста, тиреотропный гормон, уровень кальция | Нормальный гипофиз и щитовидная железа |
| Эккринные системы | Уменьшено производство пота, слезы, слюна | - |
Смотрите также
- Болезнь ускоренного старения
- Биогеронтология
- Дегенеративное заболевание
- Генетическое расстройство
- CAMFAK синдром - считается формой (или подмножеством) синдрома Кокейна[20]
Рекомендации
- ^ Бертола; Cao, H; Альбано, Lm; Oliveira, Dp; Кок, Ф; Marques-Dias, Mj; Ким, Калифорния; Hegele, Ра (2006). «Синдром Кокейна типа А: новые мутации у восьми типичных пациентов». Журнал генетики человека. 51 (8): 701–5. Дои:10.1007 / s10038-006-0011-7. PMID 16865293.
- ^ Джеймс, Уильям; Бергер, Тимоти; Элстон, Дирк (2005). Болезни кожи Эндрюса: клиническая дерматология (10-е изд.). Сондерс. п.575. ISBN 978-0-7216-2921-6.
- ^ а б c d е Бендер М., Потоцкий Л., Метри Д. Что это за синдром? Синдром Кокейна. Детская дерматология [сериал онлайн]. Ноябрь 2003 г .; 20 (6): 538-540. Доступно по адресу: MEDLINE с полным текстом, Ипсвич, Массачусетс. Доступ 30 апреля 2015 г.
- ^ Hoeijmakers JH (октябрь 2009 г.). «Повреждение ДНК, старение и рак». N. Engl. J. Med. 361 (15): 1475–85. Дои:10.1056 / NEJMra0804615. PMID 19812404.
- ^ а б c d Нэнси М., Берри С. (1 января 1992 г.). «Синдром Кокейна: обзор 140 случаев». Американский журнал медицинской генетики. 42 (1): 68–84. Дои:10.1002 / ajmg.1320420115. PMID 1308368.
- ^ а б c d Нил CA, Dingwall MM. Синдром, напоминающий прогерию: обзор двух случаев. Архив болезней детства. 1950; 25 (123): 213-223.
- ^ а б c d е ж Синдром Кокейна. Домашний справочник по генетике http://ghr.nlm.nih.gov/condition/cockayne-syndrome Опубликовано 28 апреля 2015 г. Проверено в мае 2010 г. Проверено 30 апреля 2015 г.
- ^ а б c d е ж Джавадзаде М. Синдром Кокейна. Иран Дж. Детский Neurol. Осень 2014; 8; 4 (Дополнение 1): 18-19.
- ^ Цереброокулофациоскелетный синдром 2. Онлайн-менделевское наследование у человека. https://omim.org/entry/610756. Опубликовано 12.02.2007.
- ^ Синдром Лаугеля В. Кокейна. 2000 г. 28 декабря [Обновлено 14 июня 2012 г.]. В: Pagon RA, Adam MP, Ardinger HH, et al., Редакторы. GeneReviews® [Интернет]. Сиэтл (Вашингтон): Вашингтонский университет, Сиэтл; 1993-2015 гг. Доступна с: [1]
- ^ ван Хоффен А., Натараджан А.Т., Мейн Л.В., ван Зиланд А.А., Маллендерс Л.Х., Венема Дж. (1993). «Недостаточное восстановление транскрибированной цепи активных генов в клетках синдрома Кокейна». Нуклеиновые кислоты Res. 21 (25): 5890–5. Дои:10.1093 / nar / 21.25.5890. ЧВК 310470. PMID 8290349.
- ^ а б Ияма Т., Уилсон Д.М. (2016). «Элементы, которые регулируют реакцию на повреждение ДНК белков, дефектных при синдроме Кокейна». J. Mol. Биол. 428 (1): 62–78. Дои:10.1016 / j.jmb.2015.11.020. ЧВК 4738086. PMID 26616585.
- ^ Ияма Т., Ли С.Ю., Берквист Б.Р., Гилеади О., Бор В.А., Зейдман М.М., МакХью П.Дж., Уилсон Д.М. (2015). «CSB взаимодействует с SNM1A и способствует переработке межцепочечных сшивок ДНК». Нуклеиновые кислоты Res. 43 (1): 247–58. Дои:10.1093 / нар / gku1279. ЧВК 4288174. PMID 25505141.
- ^ Батенбург Н.Л., Томпсон Е.Л., Хендриксон Е.А., Чжу XD (2015). «Белок группы В при синдроме Кокейна регулирует репарацию двухцепочечных разрывов ДНК и активацию контрольных точек». EMBO J. 34 (10): 1399–416. Дои:10.15252 / embj.201490041. ЧВК 4491999. PMID 25820262.
- ^ Вэй Л., Накадзима С., Бём С., Бернштейн К.А., Шен З., Цанг М., Левин А.С., Лан Л. (2015). «Повреждение ДНК во время фазы G0 / G1 запускает РНК-шаблонную B-зависимую гомологичную рекомбинацию с синдромом Кокейна». Proc. Natl. Акад. Sci. СОЕДИНЕННЫЕ ШТАТЫ АМЕРИКИ. 112 (27): E3495–504. Bibcode:2015PNAS..112E3495W. Дои:10.1073 / pnas.1507105112. ЧВК 4500203. PMID 26100862.
- ^ Киллермен, Мартен. Синдром Кокейна. Шведский информационный центр по редким заболеваниям. 2012: 4.0. http://www.socialstyrelsen.se/rarediseases/cockaynesyndrome#anchor_17 В архиве 2015-09-24 на Wayback Machine
- ^ Название: Синдром Кокейна Авторы: д-р Нита Р. Сутай, д-р М. Д. Ашфак Тинмасвала, д-р Манджири Карлекар, д-р Свати Джа http://jmscr.igmpublication.org/v3-i7/35%20jmscr.pdf
- ^ "Синдром Кокейна | Информационный центр по генетическим и редким заболеваниям (GARD) - программа NCATS".
- ^ Кариккинет, A.C .; Scheibye-Knudsen, M .; Fivenson, E .; Croteau, D. L .; Бор В.А. (2016). «Синдром Кокейна: клинические особенности, модельные системы и пути». Обзоры исследований старения. 33: 3–17. Дои:10.1016 / j.arr.2016.08.002. ЧВК 5195851. PMID 27507608.
- ^ «Орфанет: синдром CAMFAK».
внешняя ссылка
- Эта статья включает в себя текст из общественного достояния из Национальная медицинская библиотека США
| Классификация | |
|---|---|
| Внешние ресурсы |