Эпигенетика нейродегенеративных заболеваний - Epigenetics of neurodegenerative diseases
Эта статья нужно больше медицинские справки за проверка или слишком сильно полагается на основные источники. (Май 2015 г.) |
Нейродегенеративные заболевания представляют собой гетерогенную группу сложных расстройств, связанных с дегенерацией нейроны в любом периферическая нервная система или Центральная нервная система. Их основные причины чрезвычайно разнообразны и осложняются различными генетическими факторами и / или факторами окружающей среды. Эти заболевания вызывают прогрессирующее разрушение нейрона, что приводит к снижению преобразование сигнала а в некоторых случаях даже гибель нейронов. Заболевания периферической нервной системы можно далее классифицировать по типу нервных клеток (мотор, сенсорный, или и то, и другое), пораженных заболеванием. Эффективному лечению этих заболеваний часто препятствует отсутствие понимания основной молекулярной и генетической патологии. Эпигенетическая терапия исследуется как метод коррекции уровней экспрессии неправильно регулируемых генов при нейродегенеративных заболеваниях.
Нейроденгенеративные заболевания мотонейронов может вызвать дегенерацию моторных нейронов, участвующих в произвольном мышечном контроле, таком как сокращение и расслабление мышц. В этой статье будут рассмотрены эпигенетика и лечение бокового амиотрофического склероза (БАС) и спинальной мышечной атрофии (СМА). Увидеть Информационный бюллетень Motor Neuron для получения подробной информации о других заболеваниях двигательных нейронов. Нейродегенеративные заболевания центральной нервной системы может повлиять на мозг и / или спинной мозг. В этой статье будут рассмотрены эпигенетика и лечение Болезнь Альцгеймера (ОБЪЯВЛЕНИЕ), Болезнь Хантингтона (HD) и Болезнь Паркинсона (PD). Эти заболевания характеризуются хронической и прогрессирующей дисфункцией нейронов, иногда приводящей к поведенческим аномалиям (как при БП) и, в конечном итоге, к гибели нейронов, что приводит к слабоумие.
Нейродегенеративные заболевания сенсорных нейронов могут вызывать дегенерацию сенсорных нейронов, участвующих в передаче сенсорной информации, такой как слушание и видя. Основная группа заболеваний сенсорных нейронов - это наследственные сенсорные и вегетативные невропатии (HSAN), такие как HSAN I, HSAN II, и Шарко-Мари-Зуб Тип 2B (CMT2B).[1][2] Хотя некоторые заболевания сенсорных нейронов признаны нейродегенеративными, эпигенетические факторы в молекулярной патологии еще не выяснены.
Эпигенетики и эпигенетические препараты
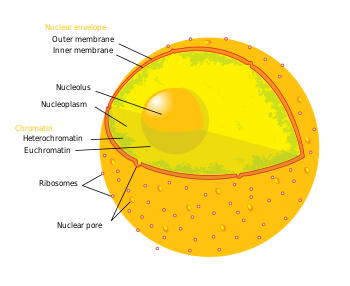
Период, термин эпигенетика относится к трем уровням генной регуляции: (1) Метилирование ДНК, (2) модификации гистонов, и (3) некодирующая РНК (нкРНК) функция. Вкратце, гистон-опосредованный контроль транскрипции происходит путем наматывания ДНК на гистон основной. Эта структура ДНК-гистонов называется нуклеосома; чем прочнее ДНК связана нуклеосомой, и чем сильнее последовательность нуклеосом сжата между собой, тем сильнее репрессивный эффект на транскрипция генов в последовательностях ДНК рядом с гистонами или обернутых вокруг них, и наоборот (т.е. более слабое связывание ДНК и ослабленное уплотнение приводят к сравнительно дерепрессированному состоянию, что приводит к факультативному гетерохроматин или, что еще более дерепрессировано, эухроматин ). В своем наиболее репрессивном состоянии, вовлекая множество складок внутрь себя и других каркасных белков, структуры ДНК-гистонов образуют конститутивный гетерохроматин. Эта структура хроматина опосредуется этими тремя уровнями регуляции генов. Наиболее важными эпигенетическими модификациями для лечения нейродегенеративных заболеваний являются метилирование ДНК и модификации гистоновых белков посредством метилирования или ацетилирования.[3][4]
- У млекопитающих метилирование происходит на ДНК и гистоновых белках. Метилирование ДНК происходит на цитозине Динуклеотиды CpG в геномной последовательности, а метилирование белка происходит на амино-концах основных белков гистонов - чаще всего на остатках лизина.[4] CpG относится к динуклеотиду, состоящему из дезоксинуклеотида цитозина, непосредственно примыкающего к дезоксинуклеотиду гуанина. Кластер динуклеотидов CpG, сгруппированных вместе, называется Остров CpG и у млекопитающих эти CpG-островки являются одним из основных классов промоторов генов, на которых или вокруг которых могут связываться факторы транскрипции и начинаться транскрипция. Метилирование динуклеотидов CpG и / или островков внутри промоторов генов связано с репрессией транскрипции через вмешательство фактор транскрипции связывание и рекрутирование транскрипционных репрессоров с помощью метильных связывающих доменов. Метилирование внутригенные области связано с повышенной транскрипцией. Группа ферментов, отвечающих за присоединение метильных групп к ДНК, называется ДНК-метилтрансферазы (ДНМТ). Фермент, отвечающий за удаление метильной группы, называется ДНК-деметилазой. Эффекты метилирование гистонов зависят от остатка (например, какая аминокислота на каком гистоновом хвосте метилирована), поэтому результирующая транскрипционная активность и регуляция хроматина может изменяться.[4] Ферменты, отвечающие за присоединение метильных групп к гистонам, называются гистоновые метилтрансферазы (HMTs). Ферменты, отвечающие за удаление метильных групп из гистона: гистоновые деметилазы.
- Ацетилирование происходит на остатках лизина, обнаруженных на N-конце аминогруппы гистоновых хвостов. Ацетилирование гистонов чаще всего связано с расслаблением хроматина, дерепрессией транскрипции и, следовательно, с активной транскрипцией генов.[4] Гистоновые ацетилтрансферазы (HAT) - это ферменты, отвечающие за добавление ацетильных групп, и гистоновые деацетилазы (HDAC) - это ферменты, отвечающие за удаление ацетильных групп. Следовательно, добавление или удаление ацетильной группы к гистону может изменить экспрессию близлежащих генов. Большинство исследуемых препаратов являются ингибиторами белков, удаляющих ацетил из гистонов или гистондеацетилаз (HDAC).
- Вкратце, нкРНК участвуют в сигнальных каскадах с эпигенетическими ферментами маркировки, такими как HMT, и / или с РНК-интерференция (RNAi) машины. Часто эти сигнальные каскады приводят к эпигенетической репрессии (например, см. Инактивация Х-хромосомы ), хотя в некоторых случаях верно обратное. Например, BACE1-AS Экспрессия нкРНК повышается у пациентов с болезнью Альцгеймера и приводит к повышению стабильности BACE1 - предшественник мРНК фермента, участвующего в болезни Альцгеймера.[5]
Эпигенетические препараты нацелены на белки, ответственные за модификации ДНК или гистонов. Современные эпигенетические препараты включают, но не ограничиваются: Ингибиторы HDAC (HDACi), модуляторы HAT, ингибиторы ДНК-метилтрансферазы и ингибиторы гистоновой деметилазы.[6][7] Большинство эпигенетических препаратов, протестированных для использования против нейродегенеративных заболеваний, являются ингибиторами HDAC; однако некоторые ингибиторы DNMT также были протестированы. В то время как большинство эпигенетических лекарств проводилось на моделях мышей, некоторые эксперименты проводились на человеческих клетках, а также в испытаниях лекарств на людях (см. Таблицу ниже). Использование эпигенетических препаратов в качестве лечения нейродегенеративных расстройств сопряжено с определенными рисками, поскольку некоторые эпигенетические препараты (например, HDAC, такие как бутират натрия ) неспецифичны по отношению к своим мишеням, что оставляет возможность для нецелевых эпигенетических меток, вызывающих нежелательные эпигенетические модификации.
| Функция | Классификация | Препарат, средство, медикамент | ALS | ОБЪЯВЛЕНИЕ | HD | PD | SMA |
|---|---|---|---|---|---|---|---|
| Ингибитор метилирования ДНК | химический аналог из цитидин | Азатиоприн | M (н.у.) | M (н.у.) | |||
| Ингибитор HDAC (малая молекула ) | бензамид | M344 | MC 19 | ||||
| жирная кислота | Бутират натрия | Мой) 5, 6, 7 ; H (н.у.) | D (г) 11 | Мой) 14; R (у) 15; D (г) 16, 18; H (н.у.) | MC 20; Мой) 21; H (н.у.) | ||
| Фенилбутират натрия | Мой) 1; H (у) 2 | Мой) 8; H (н.у.) | H (ys) 12 | MC 20; H (v) 21, 22 | |||
| Вальпроевая кислота | Мой) 2; H (ni) 3 | Мой) 9; H (н.у.) | D (г) 11 | R (у) 17; H (н.у.) | MC 23, 24; Мой) 25; H (v) 26, 27, 28, 29 | ||
| гидроксамовая кислота | Трихостатин А | Мой) 4; H (н.у.) | Мой) 10; H (н.у.) | MC 13; D (г) 11 | Мой) 30, 31; H (н.у.) | ||
| Вориностат (суберанилогидроксамовая кислота -САГА) | Мой) 9; H (н.у.) | MC 13; D (г) 11 | D (г) 18 | MC 32, 33; Мой) 34; H (н.у.) |
- Болезнь: боковой амиотрофический склероз (ALS), Болезнь Альцгеймера (ОБЪЯВЛЕНИЕ), болезнь Хантингтона (HD), спинальная мышечная атрофия (SMA), болезнь Паркинсона (PD)
- Проверено на: мыши (M), только клетках мыши (MC), человеке (H), Дрозофила (D), крыса (R)
- Успешное лечение: да (y), да, но с побочными эффектами (ys), еще нет (ny), переменная (v), без улучшения (ni)
- Рекомендации: перечисляются в столбце (болезнь) и в порядке возрастания строки (препарат)
- ALS: (1)[8][9] (2)[10] (3)[11] (4)[12]
- ОБЪЯВЛЕНИЕ: (5)[13] (6)[14] (7)[15] (8)[14] (9)[16] (10)[17]
- HD: (11)[18] (12)[19] (13)[20]
- PD: (14)[21] (15)[22] (16)[23] (17)[24] (18)[25]
- SMA: (19)[26] (20)[27] (21)[28] (22)[29] (23)[30] (24)[31] (25)[32] (26)[33] (27)[34] (28)[35] (29)[36] (30)[37] (31)[38] (32)[39] (33)[40] (34)[41]
Нейродегенеративные заболевания двигательных нейронов
Боковой амиотрофический склероз (БАС)
Боковой амиотрофический склероз (БАС), также известный как болезнь Лу Герига, представляет собой заболевание двигательных нейронов, которое связано с нейрогенерацией. Все скелетные мышцы в организме контролируются двигательными нейронами, которые передают сигналы от мозга к мышце через нервномышечное соединение. Когда двигательные нейроны дегенерируют, мышцы больше не получают сигналы от мозга и начинают истощаться. БАС характеризуется ригидностью мышц, подергиванием мышц и прогрессирующей мышечной слабостью из-за истощения мышц. Части тела, пораженные ранними симптомами БАС, зависят от того, какие двигательные нейроны в организме повреждаются в первую очередь, обычно это конечности. По мере прогрессирования заболевания большинство пациентов не могут ходить или использовать руки, и в конечном итоге у них возникают трудности с речью, глотанием и дыханием. Большинство пациентов сохраняют когнитивные функции, а сенсорные нейроны обычно не страдают. Пациентам часто ставят диагноз после 40 лет, а среднее время выживания от начала болезни до смерти составляет около 3-4 лет. На последних стадиях пациенты могут потерять произвольный контроль над глазными мышцами и часто умирают от нарушение дыхания или же пневмония в результате дегенерации двигательных нейронов и мышц, необходимых для дыхания. В настоящее время не существует лекарств от БАС, только методы лечения, которые могут продлить жизнь.
Генетика и основные причины
На сегодняшний день в БАС вовлечено множество генов и белков. Одной из общих тем для многих из этих генов и их причинных мутаций является наличие белковые агрегаты в двигательных нейронах.[42] Другие общие молекулярные особенности у пациентов с БАС - это измененный метаболизм РНК.[43] и общее гипоацетилирование гистонов.[44]
- SOD1
- В SOD1 ген на хромосома 21 который кодирует белок супероксиддисмутазы, встречается в 2% случаев и, как полагают, передается через аутосомно-доминантный манера.[45] Многие различные мутации в SOD1 были зарегистрированы у пациентов с БАС с разной степенью прогрессирования. Белок SOD1 отвечает за разрушение встречающихся в природе, но вредных супероксидные радикалы произведенный митохондрии. Большинство мутаций SOD1, связанных с БАС, представляют собой мутации с усилением функции, при которых белок сохраняет свою ферментативную активность, но агрегируется в мотонейронах, вызывая токсичность.[46][47] Нормальный белок SOD также участвует в других случаях БАС из-за потенциально клеточного стресса.[48] Была разработана мышиная модель БАС посредством мутаций с усилением функции в SOD1.[49]
- c9orf72
- Ген под названием c9orf72 было обнаружено, что в некодирующей области гена присутствует гексануклеотидный повтор в ассоциации с ALS и ALS-FTD.[50] Эти гексануклеотидные повторы могут присутствовать до 40% семейных случаев БАС и 10% спорадических случаев. C9orf72, вероятно, функционирует как фактор обмена гуанина для небольшого GTPase, но это, вероятно, не связано с основной причиной БАС.[51] Гексануклеотидные повторы, вероятно, вызывают клеточную токсичность после того, как они сращенный из транскриптов мРНК c9orf72 и накапливаются в ядрах пораженных клеток.[50]
- UBQLN2
- В UBQLN2 ген кодирует белок убихилин 2, который отвечает за контроль деградации убиквитинированный белки в клетке. Мутации в UBQLN2 мешают деградации белка, что приводит к нейродегенерации из-за аномальной агрегации белка.[52] Эта форма БАС сцеплена с Х-хромосомой и доминантно наследуется, а также может быть связана с слабоумие.
Эпигенетическое лечение ингибиторами HDAC
Пациенты с БАС и мышиные модели демонстрируют общее гипоацетилирование гистонов, которое в конечном итоге может вызвать апоптоз ячеек.[53] В экспериментах на мышах ингибиторы HDAC противодействуют этому гипоацетилированию, реактивируют аберрантно подавленные гены и противодействуют инициации апоптоза.[12][54] Кроме того, известно, что ингибиторы HDAC предотвращают образование агрегатов белка SOD1 in vitro.[55]
- Фенилбутират натрия
- Фенилбутират натрия Лечение на мышиной модели БАС SOD1 показало улучшение двигательной активности и координации, снижение нервной атрофии и невральной потери, а также увеличение веса.[8][9] Высвобождение проапоптотических факторов также было отменено, как и общее повышение ацетилирования гистонов.[54] Испытание на людях с использованием фенилбутурата у пациентов с БАС показало некоторое увеличение ацетилирования гистонов, но в исследовании не сообщалось, улучшились ли симптомы БАС при лечении.[10]
- Вальпроевая кислота
- Вальпроевая кислота в исследованиях на мышах восстановились уровни ацетилирования гистонов, повысились уровни факторов, способствующих выживанию, а у мышей улучшилась двигательная способность.[56] Однако, хотя препарат задерживал начало БАС, он не увеличивал продолжительность жизни и не предотвращал денервация.[57] Испытания на людях вальпроевой кислоты у пациентов с БАС не улучшили выживаемость или не замедлили прогрессирование.[11]
- Трихостатин А
- Трихостатин А испытания на мышиных моделях БАС восстановили ацетилирование гистонов в спинномозговых нейронах, уменьшили демиелинизацию аксонов и увеличили выживаемость мышей.[12]
Спинальная мышечная атрофия (СМА)

Спинальная мышечная атрофия (СМА) - аутосомно-рецессивное заболевание двигательных нейронов, вызванное мутациями в SMN1 ген.[58] Симптомы сильно различаются в зависимости от каждой подгруппы СМА и стадии заболевания. Общие симптомы включают общую мышечную слабость и плохой мышечный тонус, включая конечности и дыхательные мышцы, что приводит к затруднениям при ходьбе, дыхании и кормлении. В зависимости от типа СМА заболевание может проявляться от младенчества до взрослого возраста. Поскольку белок SMN обычно способствует выживанию мотонейронов, мутации в SMN1 приводят к медленной дегенерации моторных нейронов, ведущей к прогрессирующему общесистемному истощению мышц. В частности, со временем снижение уровня белка SMN приводит к постепенной гибели альфа двигательные нейроны в передний рог спинного мозга и мозг. Мышцы зависят от связей с двигательными нейронами и центральной нервной системой, чтобы стимулировать поддержание мышц, и поэтому дегенерация двигательных нейронов и последующая денервация мышц приводят к потере мышечного контроля и атрофии мышц. Часто сначала поражаются мышцы нижних конечностей, затем верхние конечности, а иногда и мышцы дыхания и жевания. Как правило, проксимальная мышца всегда поражается больше, чем дистальная.
Генетическая причина
Спинальная мышечная атрофия связана с генетическими мутациями в гене SMN1 (выживание моторного нейрона 1). Белок SMN широко экспрессируется в нейронах и выполняет множество функций внутри нейронов, включая сплайсосома конструкция, транспорт мРНК аксонов, нейрит нарост в процессе развития, и нервномышечное соединение формирование. Причинно-следственная потеря функции при СМА в настоящее время неизвестна.
SMN1 находится в теломерный регион хромосома человека 5 а также содержит SMN2 в центромерный область, край. SMN1 и SMN2 почти идентичны, за исключением одного нуклеотидная замена в SMN2, что приводит к альтернативному сайту сплайсинга, где интрон 6 встречается с экзоном 8. Это изменение единственной пары оснований приводит только к 10-20% транскриптов SMN2, приводящих к полностью функциональному белку SMN, и 80-90% транскриптов, приводящим к усеченному белку, который является быстро деградировал. У большинства пациентов с СМА имеется 2 или более копий гена SMN2 с большим количеством копий, что снижает тяжесть заболевания.[59] Большинство пациентов с СМА имеют либо точечные мутации или делеция в экзоне 7, часто приводящая к белковому продукту, подобному усеченной и деградированной версии белка SMN2. У пациентов с СМА это небольшое количество функционального белкового продукта SMN2 позволяет некоторым нейронам выжить.
Эпигенетическое лечение посредством активации гена SMN2
Хотя СМА не вызывается эпигенетическим механизмом, терапевтические препараты, нацеленные на эпигенетические метки, могут дать пациентам с СМА некоторое облегчение, остановить или даже обратить вспять прогрессирование заболевания. Поскольку пациенты с СМА с более высоким числом копий гена SMN2 имеют менее тяжелые симптомы, исследователи предсказали, что эпигенетические препараты, которые увеличивают экспрессию мРНК SMN2, увеличат количество функционального белка SMN в нейронах, что приведет к уменьшению симптомов СМА. Ингибиторы гистон-деацетилазы (HDAC) являются основными соединениями, которые были протестированы на повышение экспрессии мРНК SMN2. Ингибирование HDAC допускает гиперацетилирование локусов гена SMN2, теоретически приводящее к увеличению экспрессии SMN2.[40] Многие из этих ингибиторов HDAC (HDACi) сначала тестируются на мышиных моделях SMA, созданных посредством различных мутаций в гене SMN1 мыши. Если у мышей наблюдается улучшение и лекарство не вызывает очень много побочных эффектов или токсичности, лекарство можно использовать в клинических испытаниях на людях. Испытания на людях всех нижеперечисленных ингибиторов HDAC чрезвычайно разнообразны и часто зависят от точного подтипа СМА пациента.
- Квизиностат (JNJ-26481585)
- Quisinostat эффективен при низких дозах, что приводит к некоторому улучшению нервно-мышечной функции на мышиной модели СМА, но выживаемость не увеличивается.[60] Никаких испытаний на людях не проводилось.
- Бутират натрия
- Бутират натрия был первым ингибитором HDAC, испытанным на моделях мышей со SMA. Он продлил продолжительность жизни мышей со SMA на 35% и показал повышенный уровень белка SMN в ткани спинного мозга.[27][28] Однако до настоящего времени бутират натрия не использовался в испытаниях на людях.
- Фенилбутират натрия
- Фенилбутират натрия увеличивает количество транскриптов мРНК полной длины SMN2 в клеточной культуре, но применение лекарственного средства необходимо повторять для сохранения результатов.[27] Испытания на людях показывают смешанные результаты: одно исследование показало повышение уровня транскрипта СМА в крови и улучшение двигательной функции.[29] но более крупное испытание не показало влияния на прогрессирование заболевания или двигательную функцию.[28]
- Вальпроевая кислота
- Вальпроевая кислота добавление к клеткам пациентов с СМА увеличивало уровни мРНК и белка SMN2, и это лекарство непосредственно активирует промотор SMN2.[30][31] В модели мышей со SMA вальпроевая кислота добавлялась к питьевой воде и восстанавливала плотность мотонейронов и увеличивала количество мотонейронов в течение 8 месяцев.[32] Испытания на людях чрезвычайно разнообразны, демонстрируя повышенный уровень SMN2 и увеличенную мышечную силу в одних испытаниях и абсолютно никаких эффектов в других испытаниях.[34][33][35][36]
- M344
- M344 представляет собой бензамид, который показывает многообещающие результаты в культуре клеток фибробластов и увеличивает уровень факторов сплайсинга, которые, как известно, модулируют транскрипты SMN2, но лекарство было признано токсичным, и исследования не продвинулись до тестирования in vivo.[26]
- Трихостатин А
- Трихостатин А лечение показывает многообещающие результаты у мышей. В одном исследовании трихостатин А в сочетании с дополнительным питанием в моделях с ранним началом СМА у мышей приводил к улучшению двигательной функции и выживаемости и задерживал прогрессирующую денервацию мышц.[37] Второе исследование на мышиной модели SMA показало увеличение количества транскриптов SMN2 при ежедневных инъекциях.[38] Никаких испытаний на людях не проводилось.
- Вориностат (САХА)
- Вориностат представляет собой ингибитор второго поколения, который довольно нетоксичен и эффективен в культуре клеток при низких концентрациях.[39] и увеличивает ацетилирование гистонов на промоторе SMN2.[40] В модели мышей со SMA лечение SAHA приводило к увеличению веса, увеличению уровней транскриптов SMN2 в мышцах и спинном мозге, а также к остановке потери и денервации мотонейронов.[41] Никаких испытаний на людях не проводилось.
Нейродегенеративные заболевания центральной нервной системы
Болезнь Альцгеймера (AD)
Болезнь Альцгеймера (БА) - самая распространенная форма деменции среди пожилых людей. Заболевание с поведенческой точки зрения характеризуется хроническим прогрессирующим снижением когнитивной функции, которое начинается с кратковременной потери памяти, а с точки зрения неврологии - накоплением неправильно свернутых тау-белок и связанные нейрофибриллярные сплетения и бета-амилоидными сенильными бляшками бета-амилоид сенильные бляшки. Было установлено, что несколько генетических факторов способствуют развитию БА, включая мутации белок-предшественник амилоида (ПРИЛОЖЕНИЕ) и пресенилины 1 и 2 гены и семейное наследование аполипопротеин E аллель эпсилон 4. В дополнение к этим общим факторам, существует ряд других генов, которые показали измененную экспрессию при болезни Альцгеймера, некоторые из которых связаны с эпигенетическими факторами.
Эпигенетические факторы

- нкРНК
- нкРНК, которая кодируется антисмысловой частью интрона в гене фермента, расщепляющего бета-амилоид, BACE1, участвует в AD.[5] Эта нкРНК, BACE1-AS (для антисмысловой), который перекрывает экзон 6 BACE1, участвует в повышении устойчивости BACE1 транскрипт мРНК. Как следует из названия этого гена, BACE1 - это ферментативный белок, который расщепляет белок-предшественник амилоида до нерастворимой бета-формы амилоида, которая затем агрегируется в сенильные бляшки. С повышенной стабильностью BACE1 мРНК в результате BACE1-AS, более BACE1 мРНК доступна для трансляции в белок BACE1.
- miRNA
- не всегда было показано, что факторы играют роль в прогрессировании AD. miRNA участвуют в посттранскрипционном молчании генов посредством ингибирования трансляции или участия в РНКи пути. Некоторые исследования показали повышающую регуляцию miRNA-146a, которая по-разному регулирует экспрессию связанных с нейроиммунными интерлейкином-1R киназ IRAK1 и IRAK2 в мозге человека с БА, в то время как другие исследования показали повышенную или понижающую регуляцию miRNA-9 в головном мозге.[61]
- Метилирование ДНК
- В случаях болезни Альцгеймера наблюдалось глобальное гипометилирование ДНК и специфичное для генов гиперметилирование, хотя результаты исследований различались, особенно в исследованиях человеческого мозга. Гипотетически глобальное гипометилирование должно быть связано с глобальным увеличением транскрипции, поскольку CpG-островки наиболее распространены в промоторах генов; специфическое для генов гиперметилирование, однако, может указывать на то, что эти гиперметилированные гены репрессируются метками метилирования. Обычно репрессивное гиперметилирование генов, связанных с обучением и памятью, наблюдается в сочетании с депрессивным гипометилированием нейровоспалительных генов и генов, связанных с патологической экспрессией болезни Альцгеймера. Снижение метилирования было обнаружено в нейронах височной коры головного мозга, связанных с долговременной памятью, у монозиготных близнецов с болезнью Альцгеймера по сравнению со здоровыми близнецами.[62] Глобальное гипометилирование динуклеотидов CpG также наблюдалось в гиппокампе.[63] и во II слое энторинальной коры[64] пациентов с БА, оба из которых подвержены патологии БА. Эти результаты, полученные путем зондирования с помощью иммуноанализа, были опровергнуты исследованиями, в которых изучалась последовательность ДНК с помощью бисульфитное секвенирование, техника трансформации CpG, которая чувствительна к статусу метилирования CpG, при котором наблюдается глобальное гипометилирование.[65][66]
- СОХ-2
- На уровне отдельных генов гипометилирование и, следовательно, дерепрессия СОХ-2 происходит, ингибирование которого уменьшает воспаление и боль, и гиперметилирование BDNF, нейротрофический фактор, важный для долговременной памяти.[66] Выражение CREB, зависимый от активности фактор транскрипции, участвующий в регуляции BDNF среди многих других генов также было показано, что они гиперметилированы и, таким образом, подавлены в мозге при БА, что еще больше снижает BDNF транскрипция.[66] Более того, синаптофизин (SYP), главный ген, кодирующий белок синаптических везикул, гиперметилирован и, таким образом, подавлен, а фактор транскрипции NF-κB, который участвует в передаче иммунных сигналов, как было показано, гипометилирован и, таким образом, дерепрессирован.[66] Взятые вместе, эти результаты прояснили роль нарушения регуляции генов, участвующих в обучении, памяти и синаптической передаче, а также в иммунном ответе.
- Гипометилирование
- наблюдается в промоторах пресенилин 1,[67] GSK3beta, который фосфорилирует тау-белок,[68] и BACE1,[69] фермент, который расщепляет АРР до бета-амилоидной формы, которая, в свою очередь, объединяется в нерастворимые сенильные бляшки. Репрессивное гиперметилирование, вызванное бета-амилоидом, наблюдалось на промоторе Нэп, ген неприлизина, который является основным ферментом, очищающим бета-амилоид в головном мозге.[70] Это подавление NEP могло привести к накоплению сенильных бляшек с прямой связью; в сочетании с наблюдаемым увеличением AD мозга BACE1-AS и соответствующее увеличение белка BACE1 и бета-амилоида,[5] несколько уровней эпигенетической регуляции могут участвовать в контроле образования, клиренса или агрегации бета-амилоида, а также отложения сенильных бляшек. Возраст может иметь некоторое влияние на уровни метилирования ДНК в промоторах определенных генов, поскольку одно исследование обнаружило более высокие уровни метилирования у ПРИЛОЖЕНИЕ промоторы у пациентов с БА до 70 лет, но более низкие уровни метилирования у пациентов старше 70 лет.[71] Исследования дифференциального метилирования ДНК в мозге человека с БА остаются в основном неубедительными, возможно, из-за высокой степени вариабельности между людьми и многочисленных комбинаций факторов, которые могут привести к БА.
- Следы гистона
- Ацетилирование остатков лизина на гистоновых хвостах обычно связано с активацией транскрипции, тогда как деацетилирование связано с репрессией транскрипции. Есть несколько исследований, изучающих специфические гистоновые метки при БА. Эти исследования выяснили уменьшение ацетилирования лизинов 18 и 23 на N-концевых хвостах гистона 3 (H3K18 и H3K23, соответственно).[72] и увеличение HDAC2 в головном мозге AD[73] - оба знака относятся к репрессии транскрипции. Связанное с возрастом снижение когнитивных функций было связано с нарушением регуляции ацетилирования H4K12, когнитивного эффекта, который восстанавливается у мышей путем индукции этой метки.[74]
Лечение
Лечение для профилактики или лечения болезни Альцгеймера оказалось проблематичным, поскольку болезнь является хронической и прогрессирующей, и многие эпигенетические препараты действуют глобально, а не специфично для генов. Как и в случае других потенциальных методов лечения не допустить или же улучшать Симптомы AD, эти методы лечения не работают, чтобы вылечить, а только временно ослабляют симптомы болезни, подчеркивая хронический прогрессирующий характер AD и изменчивость метилирования в мозге AD.
- Фолиевая кислота и другие витамины группы B
- Витамины группы В участвуют в метаболическом пути, который приводит к производству SAM. SAM является донором метильной группы, используемой ДНК-метилтрансферазами (DNMT) для метилирования CpG. Используя модели на животных, Fuso et al. продемонстрировали восстановление метилирования на ранее гипометилированных промоторах пресенилин 1, BACE1 и ПРИЛОЖЕНИЕ[75] - гипотетически стабильная эпигенетическая модификация, которая должна репрессировать эти гены и замедлять прогрессирование БА. Также было показано, что пищевые добавки SAM уменьшают окислительный стресс и задерживают накопление неврологических признаков AD, таких как бета-амилоид и фосфорилированный тау-белок, у трансгенных мышей AD.
- АЗА
- Хан и его коллеги продемонстрировали потенциальную роль нейроглобинин ослабление нейротоксичности, связанной с амилоидом.[76] 5-аза-2 'дезоксицитидин (АЗА или децитабин), ингибитор DNMT, продемонстрировал некоторые доказательства регуляции экспрессии нейроглобина, хотя это открытие не было проверено на моделях БА.[77]
- Гистоновые методы лечения
- Хотя исследований гистоновых меток в головном мозге при БА немного, в нескольких исследованиях изучались эффекты HDACis при лечении болезни Альцгеймера. Ингибиторы HDAC класса I и II, такие как трихостатин А, вориностат и бутират натрия, и HDACis класса III, такие как никотинамид, были эффективны при лечении симптомов в моделях AD на животных. Хотя он является многообещающим терапевтическим средством на животных моделях, исследования долгосрочной эффективности HDACis и испытания на людях еще предстоит провести.
- Бутират натрия
- Бутират натрия относится к классу I и II HDACi, и было показано, что он восстанавливает обучение и память через 4 недели.[13] уменьшить фосфорилированный тау-белок и восстановить плотность дендритных шипов в гиппокампе трансгенных мышей с БА.[14] Ацетилирование гистонов в результате диффузного применения бутирата натрия особенно распространено в гиппокампе, а гены, участвующие в обучении и памяти, показали повышенное ацетилирование у мышей с БА, получавших это лекарство.[15]
- Трихостатин А
- Трихостатин А также относится к HDACi класса I и II, который спасает обучение страху в парадигме кондиционирования страха у трансгенных мышей с БА до уровней дикого типа посредством ацетилирования на лизиновых хвостах гистона 4.[17]
- Вориностат
- Вориностат представляет собой HDACi класса I и II, который, как было показано, особенно эффективен при ингибировании HDAC2 и восстановлении функций памяти в моделях дефицита обучения, не связанных с AD.[78] Одно исследование показало, что вориностат эффективен при устранении дефицита контекстной памяти у трансгенных мышей с БА.[16]
Хантингтона (HD)

Болезнь Хантингтона (БХ) - это наследственное заболевание, которое вызывает прогрессирующую дегенерацию нейронов в кора головного мозга и полосатое тело мозга[79] что приводит к потере двигательных функций (непроизвольные сокращения мышц), снижению когнитивных способностей (что в конечном итоге приводит к деменции) и изменениям в поведении.[6]
Генетика и основные причины
Болезнь Хантингтона вызвана аутосомно-доминантной мутацией, увеличивающей количество повторов кодона глутамина (CAG) в пределах Хантингтин ген (Htt).[79] Ген Htt кодирует белок хантингтин, который играет роль в нормальном развитии, но его точная функция остается неизвестной.[80] Длина этого повтора CAG коррелирует с возрастом начала заболевания. У среднего человека без Хантингтона в гене Htt присутствует менее 36 CAG-повторов. Когда длина повтора превышает 36, начало деградации нейронов и физические симптомы болезни Гентингтона могут варьироваться от 5 лет (повтор CAG> 70) до 80 лет (повтор CAG <39).[81]
Это расширение CAG приводит к подавлению мРНК определенных генов, снижению ацетилирования гистонов и увеличению метилирования гистонов.[82][83] Точный механизм того, как этот повтор вызывает нарушение регуляции гена, неизвестен, но модификация эпигенома может играть роль. Для раннего начала болезни Хантингтона (возраст 5-15 лет) как у трансгенных мышей, так и у линий полосатых клеток мышей наблюдается специфическое для мозга гипоацетилирование гистона H3 и сниженная ассоциация гистонов со специфическими подавляющими генами в полосатом теле (а именно Bdnf, Cnr1, Drd2 - рецептор дофамина 2 и Penk1 - препроэнкефалин).[84] Как для позднего, так и для раннего начала болезни Гентингтона, ядерные гистоны H3 и H4, связанные с этими подавленными генами у мутантов Htt, имеют гипоацетилирование (пониженное ацетилирование) по сравнению с Htt дикого типа.[83][84] Этого гипоацетилирования достаточно, чтобы вызвать более плотную упаковку хроматина и подавление мРНК.[83]
Наряду с гипоацетилированием H3 как пациенты-люди, так и мыши с мутантным Htt имеют повышенные уровни триметилирования гистона H3 лизина 9.[82] Это увеличение триметилирования H3-K9 связано с повышенной экспрессией метилтрансферазы ESET / SETDB1 (ERG-ассоциированный белок с доменом SET (ESET)), который нацелен на и триметилирует остатки H3-K9.[82] Предполагается, что это гиперметилирование может объяснять начало репрессии специфических генов у мутантов Htt.[82]
Ингибиторы HDAC
Пациенты Хантингтона, а также модели мышей и дрозофилы демонстрируют гипоацетилирование гистонов H3 и H4. В настоящее время не существует лечения этого заболевания, но были протестированы многочисленные ингибиторы HDAC, и было показано, что они обращают вспять определенные симптомы, вызванные мутацией Htt.
- Бутират натрия
- Обработка бутиратом натрия замедляла дегенерацию нейронов на моделях Drosophila.[18] Обработка бутиратом натрия также увеличивала ацетилирование гистона H3 и нормализовала уровни мРНК для мутантных генов с пониженной регуляцией Htt.[84]
- Вальпроевая кислота
- Обработка вальпроевой кислотой увеличивала уровни ацетилирования мутантного Htt H3 и H4, сравнимые с Htt дикого типа на моделях Drosophila.[18]
- Фенилбутират натрия
- Фенилбутират натрия фазы II человеческого триасла с 12-15 г / день показал восстановленные уровни мРНК репрессированных генов Htt, но также имел побочные эффекты, такие как тошнота, головные боли и нестабильность прироста.[85] Также было показано, что фенилбутират увеличивает ацетилирование гистонов, снижает метилирование гистонов, увеличивает выживаемость и снижает скорость деградации нейронов на моделях мутантных мышей Htt.[19]
- Трихостатин А
- Обработка трихостатином А (TSA) увеличивала уровни ацетилирования мутантного Htt H3 и H4, сравнимые с Htt дикого типа на моделях Drosophila.[18] Также было показано, что лечение TSA увеличивает ацетилирование альфа-тубулина лизина 40 в полосатых клетках мышей и увеличивает внутриклеточный транспорт BDNF, нейротрофического фактора головного мозга, который участвует в росте и поддержании нервов в мозге.[86][20]
- Вориностат (САХА)
- Лечение вориностатом замедляло дегенерацию фоторецепторов и увеличивало продолжительность жизни взрослых мутантных Htt дрозофил.[18] Как и TSA, лечение SAHA увеличивало ацетилирование альфа-тубулина лизина 40 в полосатых клетках мышей, а также увеличивало внутриклеточный транспорт BDNF.
Болезнь Паркинсона (БП)
Болезнь Паркинсона (БП) характеризуется прогрессирующей дегенерацией дофаминергических нейронов в черной субстанции по неизвестным причинам. Некоторые гены и факторы окружающей среды (например, воздействие пестицидов) могут играть роль в возникновении болезни Паркинсона. Признаки включают мутации гена альфа-синуклеина, SNCA, а также ПАРК2, РОЗОВЫЙ1, УЧЛ1, DJ1, и LRRK2 гены и фибриллярное накопление Тела Леви из неправильно свернутого альфа-синуклеина. Симптомы наиболее заметно проявляются в нарушениях движений, включая тряску, ригидность, дефицит контролируемых движений, а также медленную и трудную ходьбу. The late stages of the disease result in dementia and depression. Levodopa and dopaminergic therapy may ameliorate symptoms, though there is no treatment to halt progression of the disease.
Epigenetic factors
- нкРНК
- Reductions of miR-133b correlated to decreased numbers of dopaminergic neurons in the midbrain of PD patients.[87] miR-132, meanwhile, is negatively correlated with dopaminergic neuron differentiation in the midbrain.[88] miR-7 and miR-153 act to reduce alpha-synuclein levels (a hallmark of PD) but are reduced in PD brain.[89]
- Метилирование ДНК
- Neurons of PD patients show hypomethylation of tumor necrosis factor (TNF) alpha encoding sequence, overexpression of which leads to apoptosis of neurons.[90] Cerebrospinal fluid of PD patients also shows elevated TNF alpha.[91] Research indicates there may be a link between DNA methylation and SNCA expression.[92][93] Furthermore, human and mouse models have shown reduction of nuclear DNMT1 levels in PD subjects, resulting in hypomethylated states associated with transcriptional repression.[94]
- Histone marks
- alpha-synuclein, the protein encoded by SNCA, can associate with histones and prevent their acetylation in concert with the HDACs HDAC1 and Sirt2.[25][95] Furthermore, it has been demonstrated that alpha-synuclein binds histone 3 and inhibits its acetylation in Дрозофила.[25] Dopamine depletion in Parkinson’s disease is associated with repressive histone modifications, including reduced H3K4me3, and lower levels of H3 and H4 lysine acetylation after levodopa therapy (a common treatment of PD).
Лечение
Epigenetic treatments tested in models of PD are few, though some promising research has been conducted. Most treatments investigated thus far are directed at histone modifications and analysis of their roles in mediating alpha-synuclein expression and activity. Pesticides and paraquat increase histone acetylation, producing neurotoxic effects similar to those seen in PD, such as apoptosis of dopaminergic cells.[96] Despite this, treatment with HDACis[97] seems to have a neuroprotective effect.
- Бутират натрия
- Several studies using different animal models have demonstrated that sodium butyrate may be effective in reducing alpha-synuclein-related neurotoxicity.[21][22] В Дрозофила, sodium butyrate improved locomotor impairment and reduced early mortality rates.[23]
- Вальпроевая кислота
- In an inducible rat model of PD, valproic acid had a neuroprotective effect by preventing translocation of alpha-synuclein into cell nuclei.[24]
- Вориностат
- In an alpha-synuclein overexpressing Дрозофила model of PD, vorinostat (as well as sodium butyrate) reduced alpha-synuclein-mediated neurotoxicity.[25]
- siRNA inhibition of SIRT2
- Treatment with SIRT2 inhibiting siRNA leads to reduced alpha-synuclein neurotoxicity AK-1 or AGK-2.[95]
Смотрите также
Рекомендации
- ^ Онлайн-менделевское наследование в человеке (OMIM): 600882 Charcot-Marie-Tooth Disease, Axonal, Type 2B; CMT2B - 600882
- ^ Сгирланзони А., Парейсон Д., Лаурия Г. (июнь 2005 г.). «Болезни сенсорных нейронов». рассмотрение. Ланцет. Неврология. 4 (6): 349–61. Дои:10.1016 / S1474-4422 (05) 70096-X. PMID 15907739. S2CID 35053543.
- ^ Goll MG, Bestor TH (2005). "Eukaryotic cytosine methyltransferases". Ежегодный обзор биохимии. 74: 481–514. Дои:10.1146/annurev.biochem.74.010904.153721. PMID 15952895.
- ^ а б c d Bernstein BE, Meissner A, Lander ES (February 2007). "The mammalian epigenome". рассмотрение. Клетка. 128 (4): 669–81. Дои:10.1016/j.cell.2007.01.033. PMID 17320505. S2CID 2722988.
- ^ а б c Faghihi MA, Modarresi F, Khalil AM, Wood DE, Sahagan BG, Morgan TE, Finch CE, St Laurent G, Kenny PJ, Wahlestedt C (July 2008). «Экспрессия некодирующей РНК повышена при болезни Альцгеймера и способствует быстрой упреждающей регуляции бета-секретазы». начальный. Природа Медицина. 14 (7): 723–30. Дои:10,1038 / нм 1784. ЧВК 2826895. PMID 18587408.
- ^ а б Urdinguio RG, Sanchez-Mut JV, Esteller M (November 2009). "Epigenetic mechanisms in neurological diseases: genes, syndromes, and therapies". Ланцет. Неврология. 8 (11): 1056–72. Дои:10.1016/S1474-4422(09)70262-5. PMID 19833297. S2CID 25946604.
- ^ Peedicayil J (April 2013). "Epigenetic drugs for Alzheimer's disease". Британский журнал клинической фармакологии. 75 (4): 1152–3. Дои:10.1111/j.1365-2125.2012.04444.x. ЧВК 3612735. PMID 22905989.
- ^ а б Del Signore SJ, Amante DJ, Kim J, Stack EC, Goodrich S, Cormier K, Smith K, Cudkowicz ME, Ferrante RJ (April 2009). "Combined riluzole and sodium phenylbutyrate therapy in transgenic amyotrophic lateral sclerosis mice". начальный. Боковой амиотрофический склероз. 10 (2): 85–94. Дои:10.1080/17482960802226148. PMID 18618304. S2CID 24124109.
- ^ а б Petri S, Kiaei M, Kipiani K, Chen J, Calingasan NY, Crow JP, Beal MF (April 2006). "Additive neuroprotective effects of a histone deacetylase inhibitor and a catalytic antioxidant in a transgenic mouse model of amyotrophic lateral sclerosis". Нейробиология болезней. 22 (1): 40–9. Дои:10.1016/j.nbd.2005.09.013. PMID 16289867. S2CID 22794616.
- ^ а б Cudkowicz ME, Andres PL, Macdonald SA, Bedlack RS, Choudry R, Brown RH, Zhang H, Schoenfeld DA, Shefner J, Matson S, Matson WR, Ferrante RJ (April 2009). "Phase 2 study of sodium phenylbutyrate in ALS". начальный. Боковой амиотрофический склероз. 10 (2): 99–106. Дои:10.1080/17482960802320487. PMID 18688762. S2CID 12390136.
- ^ а б Piepers S, Veldink JH, de Jong SW, van der Tweel I, van der Pol WL, Uijtendaal EV, Schelhaas HJ, Scheffer H, de Visser M, de Jong JM, Wokke JH, Groeneveld GJ, van den Berg LH (August 2009). "Randomized sequential trial of valproic acid in amyotrophic lateral sclerosis". начальный. Анналы неврологии. 66 (2): 227–34. Дои:10.1002/ana.21620. PMID 19743466. S2CID 44949619.
- ^ а б c Yoo YE, Ko CP (September 2011). "Treatment with trichostatin A initiated after disease onset delays disease progression and increases survival in a mouse model of amyotrophic lateral sclerosis". начальный. Экспериментальная неврология. 231 (1): 147–59. Дои:10.1016/j.expneurol.2011.06.003. PMID 21712032. S2CID 42608157.
- ^ а б Fischer A, Sananbenesi F, Wang X, Dobbin M, Tsai LH (May 2007). «Восстановление обучения и памяти связано с ремоделированием хроматина». начальный. Природа. 447 (7141): 178–82. Bibcode:2007Natur.447..178F. Дои:10.1038 / природа05772. PMID 17468743. S2CID 36395789.
- ^ а б c Ricobaraza A, Cuadrado-Tejedor M, Marco S, Pérez-Otaño I, García-Osta A (May 2012). "Phenylbutyrate rescues dendritic spine loss associated with memory deficits in a mouse model of Alzheimer disease". начальный. Гиппокамп. 22 (5): 1040–50. Дои:10.1002/hipo.20883. PMID 21069780.
- ^ а б Govindarajan N, Agis-Balboa RC, Walter J, Sananbenesi F, Fischer A (2011). "Sodium butyrate improves memory function in an Alzheimer's disease mouse model when administered at an advanced stage of disease progression". начальный. Журнал болезни Альцгеймера. 26 (1): 187–97. Дои:10.3233/JAD-2011-110080. PMID 21593570.
- ^ а б Kilgore M, Miller CA, Fass DM, Hennig KM, Haggarty SJ, Sweatt JD, Rumbaugh G (March 2010). "Inhibitors of class 1 histone deacetylases reverse contextual memory deficits in a mouse model of Alzheimer's disease". начальный. Нейропсихофармакология. 35 (4): 870–80. Дои:10.1038/npp.2009.197. ЧВК 3055373. PMID 20010553.
- ^ а б Francis YI, Fà M, Ashraf H, Zhang H, Staniszewski A, Latchman DS, Arancio O (2009). "Dysregulation of histone acetylation in the APP/PS1 mouse model of Alzheimer's disease". Журнал болезни Альцгеймера. 18 (1): 131–9. Дои:10.3233/JAD-2009-1134. PMID 19625751.
- ^ а б c d е Steffan JS, Bodai L, Pallos J, Poelman M, McCampbell A, Apostol BL, Kazantsev A, Schmidt E, Zhu YZ, Greenwald M, Kurokawa R, Housman DE, Jackson GR, Marsh JL, Thompson LM (October 2001). "Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila". начальный. Природа. 413 (6857): 739–43. Bibcode:2001Natur.413..739S. Дои:10.1038/35099568. PMID 11607033. S2CID 4419980.
- ^ а б Gardian G, Browne SE, Choi DK, Klivenyi P, Gregorio J, Kubilus JK, Ryu H, Langley B, Ratan RR, Ferrante RJ, Beal MF (January 2005). "Neuroprotective effects of phenylbutyrate in the N171-82Q transgenic mouse model of Huntington's disease". начальный. Журнал биологической химии. 280 (1): 556–63. Дои:10.1074/jbc.M410210200. PMID 15494404.
- ^ а б Dompierre JP, Godin JD, Charrin BC, Cordelières FP, King SJ, Humbert S, Saudou F (March 2007). "Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington's disease by increasing tubulin acetylation". начальный. Журнал неврологии. 27 (13): 3571–83. Дои:10.1523/JNEUROSCI.0037-07.2007. ЧВК 6672116. PMID 17392473.
- ^ а б Zhou W, Bercury K, Cummiskey J, Luong N, Lebin J, Freed CR (April 2011). "Phenylbutyrate up-regulates the DJ-1 protein and protects neurons in cell culture and in animal models of Parkinson disease". начальный. Журнал биологической химии. 286 (17): 14941–51. Дои:10.1074/jbc.M110.211029. ЧВК 3083206. PMID 21372141.
- ^ а б Rane P, Shields J, Heffernan M, Guo Y, Akbarian S, King JA (June 2012). "The histone deacetylase inhibitor, sodium butyrate, alleviates cognitive deficits in pre-motor stage PD". начальный. Нейрофармакология. 62 (7): 2409–12. Дои:10.1016/j.neuropharm.2012.01.026. PMID 22353286. S2CID 23078279.
- ^ а б St Laurent R, O'Brien LM, Ahmad ST (August 2013). "Sodium butyrate improves locomotor impairment and early mortality in a rotenone-induced Drosophila model of Parkinson's disease". начальный. Неврология. 246: 382–90. Дои:10.1016/j.neuroscience.2013.04.037. ЧВК 3721507. PMID 23623990.
- ^ а б Monti B, Gatta V, Piretti F, Raffaelli SS, Virgili M, Contestabile A (February 2010). "Valproic acid is neuroprotective in the rotenone rat model of Parkinson's disease: involvement of alpha-synuclein". начальный. Исследования нейротоксичности. 17 (2): 130–41. Дои:10.1007/s12640-009-9090-5. PMID 19626387. S2CID 40159513.
- ^ а б c d Kontopoulos E, Parvin JD, Feany MB (October 2006). "Alpha-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity". начальный. Молекулярная генетика человека. 15 (20): 3012–23. Дои:10.1093/hmg/ddl243. PMID 16959795.
- ^ а б Riessland M, Brichta L, Hahnen E, Wirth B (август 2006 г.). «Бензамид M344, новый ингибитор гистондеацетилазы, значительно увеличивает уровни РНК / белка SMN2 в клетках с мышечной атрофией спинного мозга». начальный. Генетика человека. 120 (1): 101–10. Дои:10.1007 / s00439-006-0186-1. PMID 16724231. S2CID 24804136.
- ^ а б c Андреасси С., Анджелоцци С., Тициано Ф. Д., Витали Т., Де Винченци Е., Бонинсенья А., Вилланова М., Бертини Е., Пини А., Нери Дж., Браге С. (январь 2004 г.). «Фенилбутират увеличивает экспрессию SMN in vitro: актуальность для лечения спинальной мышечной атрофии». Европейский журнал генетики человека. 12 (1): 59–65. Дои:10.1038 / sj.ejhg.5201102. PMID 14560316.
- ^ а б c Меркури Э, Бертини Э, Мессина С, Солари А, Д'Амико А, Анджелоцци С., Баттини Р., Берардинелли А, Боффи П, Бруно С., Чини С, Колитто Ф, Кинали М, Минетти С, Монджини Т, Моранди Л., Нери Дж., Орчези С., Пейн М, Пелличчони М., Пини А., Тициано Ф. Д., Вилланова М., Вита Дж., Браге С. (январь 2007 г.). «Рандомизированное двойное слепое плацебо-контролируемое испытание фенилбутирата при спинальной мышечной атрофии». начальный. Неврология. 68 (1): 51–5. Дои:10.1212 / 01.wnl.0000249142.82285.d6. PMID 17082463. S2CID 30429093.
- ^ а б Браге С., Виталий Т., Тициано Ф.Д., Анджелоцци С., Пинто А.М., Борго Ф., Москато У., Бертини Э., Меркури Э., Нери Дж. (Февраль 2005 г.). «Фенилбутират увеличивает экспрессию гена SMN у пациентов с спинальной мышечной атрофией». начальный. Европейский журнал генетики человека. 13 (2): 256–9. Дои:10.1038 / sj.ejhg.5201320. PMID 15523494.
- ^ а б Sumner CJ, Huynh TN, Markowitz JA, Perhac JS, Hill B, Coovert DD, Schussler K, Chen X, Jarecki J, Burghes AH, Taylor JP, Fischbeck KH (November 2003). "Valproic acid increases SMN levels in spinal muscular atrophy patient cells". начальный. Анналы неврологии. 54 (5): 647–54. Дои:10.1002/ana.10743. PMID 14595654. S2CID 7983521.
- ^ а б Брихта Л., Хофманн Ю., Ханен Э., Зибзехнрубль Ф.А., Рашке Х., Блюмке И., Эйюпоглу И. Ю., Вирт Б. (октябрь 2003 г.). «Вальпроевая кислота увеличивает уровень белка SMN2: препарат, хорошо известный как потенциальное средство для лечения спинальной мышечной атрофии». начальный. Молекулярная генетика человека. 12 (19): 2481–9. Дои:10.1093 / hmg / ddg256. PMID 12915451.
- ^ а б Tsai LK, Tsai MS, Lin TB, Hwu WL, Li H (November 2006). "Establishing a standardized therapeutic testing protocol for spinal muscular atrophy". начальный. Нейробиология болезней. 24 (2): 286–95. Дои:10.1016/j.nbd.2006.07.004. PMID 16952456. S2CID 31974628.
- ^ а б Weihl CC, Connolly AM, Pestronk A (August 2006). "Valproate may improve strength and function in patients with type III/IV spinal muscle atrophy". начальный. Неврология. 67 (3): 500–1. Дои:10.1212/01.wnl.0000231139.26253.d0. PMID 16775228. S2CID 13138072.
- ^ а б Piepers S, Cobben JM, Sodaar P, Jansen MD, Wadman RI, Meester-Delver A, Poll-The BT, Lemmink HH, Wokke JH, van der Pol WL, van den Berg LH (August 2011). "Quantification of SMN protein in leucocytes from spinal muscular atrophy patients: effects of treatment with valproic acid". начальный. Журнал неврологии, нейрохирургии и психиатрии. 82 (8): 850–2. Дои:10.1136/jnnp.2009.200253. PMID 20551479. S2CID 27844635.
- ^ а б Swoboda KJ, Scott CB, Crawford TO, Simard LR, Reyna SP, Krosschell KJ, Acsadi G, Elsheik B, Schroth MK, D'Anjou G, LaSalle B, Prior TW, Sorenson SL, Maczulski JA, Bromberg MB, Chan GM, Kissel JT (August 2010). «Исследование SMA CARNI-VAL, часть I: двойное слепое рандомизированное плацебо-контролируемое испытание L-карнитина и вальпроевой кислоты при спинальной мышечной атрофии». начальный. PLOS ONE. 5 (8): e12140. Bibcode:2010PLoSO ... 512140S. Дои:10.1371 / journal.pone.0012140. ЧВК 2924376. PMID 20808854.
- ^ а б Дарбар И.А., Плаггерт П.Г., Резенде МБ, Занотели Э., Рид, Калифорния (март 2011 г.). «Оценка силы мышц и двигательных способностей у детей с атрофией мышц позвоночника II и III типов, получавших вальпроевую кислоту». начальный. BMC Neurology. 11: 36. Дои:10.1186/1471-2377-11-36. ЧВК 3078847. PMID 21435220.
- ^ а б Narver HL, Kong L, Burnett BG, Choe DW, Bosch-Marcé M, Taye AA, Eckhaus MA, Sumner CJ (октябрь 2008 г.). «Устойчивое улучшение спинальной мышечной атрофии у мышей, получавших трихостатин А плюс питание». начальный. Анналы неврологии. 64 (4): 465–70. Дои:10.1002 / ana.21449. PMID 18661558. S2CID 5595968.
- ^ а б Авила А.М., Бернетт Б.Г., Тайе А.А., Габанелла Ф., Найт М.А., Хартенштейн П., Джизман З., Ди Просперо Н.А., Пеллиццони Л., Фишбек К.Х., Самнер С.Дж. (март 2007 г.). «Трихостатин А увеличивает экспрессию SMN и выживаемость в мышиной модели спинальной мышечной атрофии». начальный. Журнал клинических исследований. 117 (3): 659–71. Дои:10.1172 / JCI29562. ЧВК 1797603. PMID 17318264.
- ^ а б Hahnen E, Eyüpoglu IY, Brichta L, Haastert K, Tränkle C, Siebzehnrübl FA, Riessland M, Hölker I, Claus P, Romstöck J, Buslei R, Wirth B, Blümcke I (July 2006). "In vitro and ex vivo evaluation of second-generation histone deacetylase inhibitors for the treatment of spinal muscular atrophy". начальный. Журнал нейрохимии. 98 (1): 193–202. Дои:10.1111/j.1471-4159.2006.03868.x. PMID 16805808.
- ^ а б c Kernochan LE, Russo ML, Woodling NS, Huynh TN, Avila AM, Fischbeck KH, Sumner CJ (May 2005). "The role of histone acetylation in SMN gene expression". начальный. Молекулярная генетика человека. 14 (9): 1171–82. Дои:10.1093/hmg/ddi130. PMID 15772088.
- ^ а б Riessland M, Ackermann B, Förster A, Jakubik M, Hauke J, Garbes L, Fritzsche I, Mende Y, Blumcke I, Hahnen E, Wirth B (апрель 2010 г.). «SAHA улучшает фенотип SMA в двух моделях спинальной мышечной атрофии на мышах». начальный. Молекулярная генетика человека. 19 (8): 1492–506. Дои:10.1093 / hmg / ddq023. PMID 20097677.
- ^ Dewey CM, Cenik B, Sephton CF, Johnson BA, Herz J, Yu G (June 2012). "TDP-43 aggregation in neurodegeneration: are stress granules the key?". рассмотрение. Исследование мозга. 1462: 16–25. Дои:10.1016/j.brainres.2012.02.032. ЧВК 3372581. PMID 22405725.
- ^ Polymenidou M, Lagier-Tourenne C, Hutt KR, Bennett CF, Cleveland DW, Yeo GW (June 2012). "Misregulated RNA processing in amyotrophic lateral sclerosis". рассмотрение. Исследование мозга. 1462: 3–15. Дои:10.1016/j.brainres.2012.02.059. ЧВК 3707312. PMID 22444279.
- ^ Rouaux C, Jokic N, Mbebi C, Boutillier S, Loeffler JP, Boutillier AL (December 2003). "Critical loss of CBP/p300 histone acetylase activity by caspase-6 during neurodegeneration". начальный. Журнал EMBO. 22 (24): 6537–49. Дои:10.1093/emboj/cdg615. ЧВК 291810. PMID 14657026.
- ^ Battistini S, Ricci C, Lotti EM, Benigni M, Gagliardi S, Zucco R, Bondavalli M, Marcello N, Ceroni M, Cereda C (June 2010). "Severe familial ALS with a novel exon 4 mutation (L106F) in the SOD1 gene". начальный. Журнал неврологических наук. 293 (1–2): 112–5. Дои:10.1016/j.jns.2010.03.009. PMID 20385392. S2CID 24895265.
- ^ Bruijn LI, Houseweart MK, Kato S, Anderson KL, Anderson SD, Ohama E, Reaume AG, Scott RW, Cleveland DW (September 1998). "Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1". начальный. Наука. 281 (5384): 1851–4. Bibcode:1998Sci...281.1851B. Дои:10.1126/science.281.5384.1851. PMID 9743498.
- ^ Furukawa Y, Fu R, Deng HX, Siddique T, O'Halloran TV (May 2006). "Disulfide cross-linked protein represents a significant fraction of ALS-associated Cu, Zn-superoxide dismutase aggregates in spinal cords of model mice". начальный. Труды Национальной академии наук Соединенных Штатов Америки. 103 (18): 7148–53. Bibcode:2006PNAS..103.7148F. Дои:10.1073/pnas.0602048103. ЧВК 1447524. PMID 16636274.
- ^ Boillée S, Vande Velde C, Cleveland DW (October 2006). "ALS: a disease of motor neurons and their nonneuronal neighbors". рассмотрение. Нейрон. 52 (1): 39–59. Дои:10.1016/j.neuron.2006.09.018. PMID 17015226. S2CID 12968143.
- ^ Cudkowicz ME, McKenna-Yasek D, Sapp PE, Chin W, Geller B, Hayden DL, Schoenfeld DA, Hosler BA, Horvitz HR, Brown RH (February 1997). "Epidemiology of mutations in superoxide dismutase in amyotrophic lateral sclerosis". начальный. Анналы неврологии. 41 (2): 210–21. Дои:10.1002/ana.410410212. PMID 9029070. S2CID 25595595.
- ^ а б Todd TW, Petrucelli L (August 2016). "Insights into the pathogenic mechanisms of Chromosome 9 open reading frame 72 (C9orf72) repeat expansions". рассмотрение. Журнал нейрохимии. 138 Suppl 1: 145–62. Дои:10.1111/jnc.13623. PMID 27016280.
- ^ Yoshimura S, Gerondopoulos A, Linford A, Rigden DJ, Barr FA (October 2010). "Family-wide characterization of the DENN domain Rab GDP-GTP exchange factors". начальный. Журнал клеточной биологии. 191 (2): 367–81. Дои:10.1083/jcb.201008051. ЧВК 2958468. PMID 20937701.
- ^ Deng HX, Chen W, Hong ST, Boycott KM, Gorrie GH, Siddique N, et al. (Август 2011 г.). "Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia". начальный. Природа. 477 (7363): 211–5. Bibcode:2011Natur.477..211D. Дои:10.1038/nature10353. ЧВК 3169705. PMID 21857683.
- ^ Rouaux C, Loeffler JP, Boutillier AL (September 2004). "Targeting CREB-binding protein (CBP) loss of function as a therapeutic strategy in neurological disorders". рассмотрение. Биохимическая фармакология. 68 (6): 1157–64. Дои:10.1016/j.bcp.2004.05.035. PMID 15313413.
- ^ а б Ryu H, Smith K, Camelo SI, Carreras I, Lee J, Iglesias AH, Dangond F, Cormier KA, Cudkowicz ME, Brown RH, Ferrante RJ (June 2005). "Sodium phenylbutyrate prolongs survival and regulates expression of anti-apoptotic genes in transgenic amyotrophic lateral sclerosis mice". начальный. Журнал нейрохимии. 93 (5): 1087–98. Дои:10.1111/j.1471-4159.2005.03077.x. PMID 15934930.
- ^ Corcoran LJ, Mitchison TJ, Liu Q (March 2004). "A novel action of histone deacetylase inhibitors in a protein aggresome disease model". начальный. Текущая биология. 14 (6): 488–92. Дои:10.1016/j.cub.2004.03.003. PMID 15043813. S2CID 6465499.
- ^ Crochemore C, Virgili M, Bonamassa B, Canistro D, Pena-Altamira E, Paolini M, Contestabile A (April 2009). "Long-term dietary administration of valproic acid does not affect, while retinoic acid decreases, the lifespan of G93A mice, a model for amyotrophic lateral sclerosis". начальный. Мышцы и нервы. 39 (4): 548–52. Дои:10.1002/mus.21260. PMID 19296491.
- ^ Rouaux C, Panteleeva I, René F, Gonzalez de Aguilar JL, Echaniz-Laguna A, Dupuis L, Menger Y, Boutillier AL, Loeffler JP (May 2007). "Sodium valproate exerts neuroprotective effects in vivo through CREB-binding protein-dependent mechanisms but does not improve survival in an amyotrophic lateral sclerosis mouse model". начальный. Журнал неврологии. 27 (21): 5535–45. Дои:10.1523/JNEUROSCI.1139-07.2007. ЧВК 6672753. PMID 17522299.
- ^ Бжустович Л.М., Ленер Т., Кастилья Л.Х., Пенчасзаде Г.К., Вильгельмсен К.С., Дэниэлс Р., Дэвис К.Э., Лепперт М., Зитер Ф., Вуд Д. (апрель 1990 г.). «Генетическое картирование хронической мышечной атрофии позвоночника в детстве по хромосоме 5q11.2-13.3». начальный. Природа. 344 (6266): 540–1. Bibcode:1990Натура.344..540Б. Дои:10.1038 / 344540a0. PMID 2320125. S2CID 4259327.
- ^ Prior TW, Krainer AR, Hua Y, Swoboda KJ, Snyder PC, Bridgeman SJ, Burghes AH, Kissel JT (September 2009). "A positive modifier of spinal muscular atrophy in the SMN2 gene". начальный. Американский журнал генетики человека. 85 (3): 408–13. Дои:10.1016/j.ajhg.2009.08.002. ЧВК 2771537. PMID 19716110.
- ^ Schreml J, Riessland M, Paterno M, Garbes L, Roßbach K, Ackermann B, Krämer J, Somers E, Parson SH, Heller R, Berkessel A, Sterner-Kock A, Wirth B (June 2013). "Severe SMA mice show organ impairment that cannot be rescued by therapy with the HDACi JNJ-26481585". начальный. Европейский журнал генетики человека. 21 (6): 643–52. Дои:10.1038/ejhg.2012.222. ЧВК 3658191. PMID 23073311.
- ^ Bennett DA, Yu L, Yang J, Srivastava GP, Aubin C, De Jager PL (January 2015). "Epigenomics of Alzheimer's disease". рассмотрение. Трансляционные исследования. 165 (1): 200–20. Дои:10.1016/j.trsl.2014.05.006. ЧВК 4233194. PMID 24905038.
- ^ Mastroeni D, McKee A, Grover A, Rogers J, Coleman PD (August 2009). "Epigenetic differences in cortical neurons from a pair of monozygotic twins discordant for Alzheimer's disease". начальный. PLOS ONE. 4 (8): e6617. Bibcode:2009PLoSO...4.6617M. Дои:10.1371/journal.pone.0006617. ЧВК 2719870. PMID 19672297.
- ^ Chouliaras L, Mastroeni D, Delvaux E, Grover A, Kenis G, Hof PR, Steinbusch HW, Coleman PD, Rutten BP, van den Hove DL (September 2013). "Consistent decrease in global DNA methylation and hydroxymethylation in the hippocampus of Alzheimer's disease patients". начальный. Нейробиология старения. 34 (9): 2091–9. Дои:10.1016/j.neurobiolaging.2013.02.021. ЧВК 3955118. PMID 23582657.
- ^ Mastroeni D, Grover A, Delvaux E, Whiteside C, Coleman PD, Rogers J (December 2010). "Epigenetic changes in Alzheimer's disease: decrements in DNA methylation". начальный. Нейробиология старения. 31 (12): 2025–37. Дои:10.1016/j.neurobiolaging.2008.12.005. ЧВК 2962691. PMID 19117641.
- ^ Bakulski KM, Dolinoy DC, Sartor MA, Paulson HL, Konen JR, Lieberman AP, Albin RL, Hu H, Rozek LS (2012). "Genome-wide DNA methylation differences between late-onset Alzheimer's disease and cognitively normal controls in human frontal cortex". Журнал болезни Альцгеймера. 29 (3): 571–88. Дои:10.3233/JAD-2012-111223. ЧВК 3652332. PMID 22451312.
- ^ а б c d Rao JS, Keleshian VL, Klein S, Rapoport SI (July 2012). "Epigenetic modifications in frontal cortex from Alzheimer's disease and bipolar disorder patients". начальный. Трансляционная психиатрия. 2 (7): e132. Дои:10.1038/tp.2012.55. ЧВК 3410632. PMID 22760556.
- ^ Wang Y, Zhang JX, Du XX, Zhao L, Tian Q, Zhu LQ, Wang SH, Wang JZ (September 2008). "Temporal correlation of the memory deficit with Alzheimer-like lesions induced by activation of glycogen synthase kinase-3". Журнал нейрохимии. 106 (6): 2364–74. Дои:10.1111/j.1471-4159.2008.05578.x. PMID 18643871.
- ^ Nicolia V, Fuso A, Cavallaro RA, Di Luzio A, Scarpa S (2010). "B vitamin deficiency promotes tau phosphorylation through regulation of GSK3beta and PP2A". начальный. Журнал болезни Альцгеймера. 19 (3): 895–907. Дои:10.3233/JAD-2010-1284. PMID 20157245.
- ^ Byun CJ, Seo J, Jo SA, Park YJ, Klug M, Rehli M, Park MH, Jo I (January 2012). "DNA methylation of the 5'-untranslated region at +298 and +351 represses BACE1 expression in mouse BV-2 microglial cells". начальный. Сообщения о биохимических и биофизических исследованиях. 417 (1): 387–92. Дои:10.1016/j.bbrc.2011.11.123. PMID 22166205.
- ^ Chen KL, Wang SS, Yang YY, Yuan RY, Chen RM, Hu CJ (January 2009). "The epigenetic effects of amyloid-beta(1-40) on global DNA and neprilysin genes in murine cerebral endothelial cells". начальный. Сообщения о биохимических и биофизических исследованиях. 378 (1): 57–61. Дои:10.1016/j.bbrc.2008.10.173. PMID 19007750.
- ^ Tohgi H, Abe T, Yamazaki K, Murata T, Ishizaki E, Isobe C (July 1999). "Alterations of 3-nitrotyrosine concentration in the cerebrospinal fluid during aging and in patients with Alzheimer's disease". начальный. Письма о неврологии. 269 (1): 52–4. Дои:10.1016/S0304-3940(99)00406-1. PMID 10821643. S2CID 20536297.
- ^ Zhang K, Schrag M, Crofton A, Trivedi R, Vinters H, Kirsch W (April 2012). "Targeted proteomics for quantification of histone acetylation in Alzheimer's disease". начальный. Протеомика. 12 (8): 1261–8. Дои:10.1002/pmic.201200010. ЧВК 6812507. PMID 22577027.
- ^ Gräff J, Rei D, Guan JS, Wang WY, Seo J, Hennig KM, Nieland TJ, Fass DM, Kao PF, Kahn M, Su SC, Samiei A, Joseph N, Haggarty SJ, Delalle I, Tsai LH (February 2012). «Эпигенетическая блокада когнитивных функций в нейродегенеративном мозге». начальный. Природа. 483 (7388): 222–6. Bibcode:2012Natur.483..222G. Дои:10.1038 / природа10849. ЧВК 3498952. PMID 22388814.
- ^ Peleg S, Sananbenesi F, Zovoilis A, Burkhardt S, Bahari-Javan S, Agis-Balboa RC, Cota P, Wittnam JL, Gogol-Doering A, Opitz L, Salinas-Riester G, Dettenhofer M, Kang H, Farinelli L, Chen W, Fischer A (May 2010). "Altered histone acetylation is associated with age-dependent memory impairment in mice". начальный. Наука. 328 (5979): 753–6. Bibcode:2010Sci...328..753P. Дои:10.1126/science.1186088. PMID 20448184. S2CID 7370920.
- ^ Fuso A (March 2013). "The 'golden age' of DNA methylation in neurodegenerative diseases". рассмотрение. Clinical Chemistry and Laboratory Medicine. 51 (3): 523–34. Дои:10.1515/cclm-2012-0618. PMID 23183753. S2CID 36486849.
- ^ Khan AA, Mao XO, Banwait S, Jin K, Greenberg DA (November 2007). "Neuroglobin attenuates beta-amyloid neurotoxicity in vitro and transgenic Alzheimer phenotype in vivo". начальный. Труды Национальной академии наук Соединенных Штатов Америки. 104 (48): 19114–9. Bibcode:2007PNAS..10419114K. Дои:10.1073/pnas.0706167104. ЧВК 2141917. PMID 18025470.
- ^ Zhang W, Tian Z, Sha S, Cheng LY, Philipsen S, Tan-Un KC (2011). "Functional and sequence analysis of human neuroglobin gene promoter region". начальный. Biochimica et Biophysica Acta (BBA) - механизмы регуляции генов. 1809 (4–6): 236–44. Дои:10.1016/j.bbagrm.2011.02.003. PMID 21362510.
- ^ Guan JS, Haggarty SJ, Giacometti E, Dannenberg JH, Joseph N, Gao J, Nieland TJ, Zhou Y, Wang X, Mazitschek R, Bradner JE, DePinho RA, Jaenisch R, Tsai LH (May 2009). "HDAC2 negatively regulates memory formation and synaptic plasticity". начальный. Природа. 459 (7243): 55–60. Bibcode:2009Natur.459...55G. Дои:10.1038/nature07925. ЧВК 3498958. PMID 19424149.
- ^ а б Онлайн-менделевское наследование в человеке (OMIM): Huntington Disease - 143100
- ^ Nasir J, Floresco SB, O'Kusky JR, Diewert VM, Richman JM, Zeisler J, Borowski A, Marth JD, Phillips AG, Hayden MR (June 1995). "Targeted disruption of the Huntington's disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes". начальный. Клетка. 81 (5): 811–23. Дои:10.1016/0092-8674(95)90542-1. PMID 7774020. S2CID 16835259.
- ^ Chen S, Ferrone FA, Wetzel R (September 2002). "Huntington's disease age-of-onset linked to polyglutamine aggregation nucleation". начальный. Труды Национальной академии наук Соединенных Штатов Америки. 99 (18): 11884–9. Bibcode:2002PNAS...9911884C. Дои:10.1073/pnas.182276099. ЧВК 129363. PMID 12186976.
- ^ а б c d Ryu H, Lee J, Hagerty SW, Soh BY, McAlpin SE, Cormier KA, Smith KM, Ferrante RJ (December 2006). "ESET/SETDB1 gene expression and histone H3 (K9) trimethylation in Huntington's disease". начальный. Труды Национальной академии наук Соединенных Штатов Америки. 103 (50): 19176–81. Bibcode:2006PNAS..10319176R. Дои:10.1073/pnas.0606373103. ЧВК 1748195. PMID 17142323.
- ^ а б c Hazeki N, Tsukamoto T, Yazawa I, Koyama M, Hattori S, Someki I, Iwatsubo T, Nakamura K, Goto J, Kanazawa I (June 2002). "Ultrastructure of nuclear aggregates formed by expressing an expanded polyglutamine". начальный. Сообщения о биохимических и биофизических исследованиях. 294 (2): 429–40. Дои:10.1016/S0006-291X(02)00498-9. PMID 12051730.
- ^ а б c Sadri-Vakili G, Bouzou B, Benn CL, Kim MO, Chawla P, Overland RP, Glajch KE, Xia E, Qiu Z, Hersch SM, Clark TW, Yohrling GJ, Cha JH (June 2007). "Histones associated with downregulated genes are hypo-acetylated in Huntington's disease models". начальный. Молекулярная генетика человека. 16 (11): 1293–306. Дои:10.1093/hmg/ddm078. PMID 17409194.
- ^ Hogarth P, Lovrecic L, Krainc D (October 2007). "Sodium phenylbutyrate in Huntington's disease: a dose-finding study". начальный. Двигательные расстройства. 22 (13): 1962–4. Дои:10.1002/mds.21632. PMID 17702032.
- ^ Entrez Gene. "BDNF". United States National Center for Biotechnology Information.
- ^ Kim J, Inoue K, Ishii J, Vanti WB, Voronov SV, Murchison E, Hannon G, Abeliovich A (August 2007). "A MicroRNA feedback circuit in midbrain dopamine neurons". начальный. Наука. 317 (5842): 1220–4. Bibcode:2007Sci...317.1220K. Дои:10.1126/science.1140481. ЧВК 2782470. PMID 17761882.
- ^ Jankovic J, Chen S, Le WD (2005). "The role of Nurr1 in the development of dopaminergic neurons and Parkinson's disease". рассмотрение. Прогресс в нейробиологии. 77 (1–2): 128–38. Дои:10.1016/j.pneurobio.2005.09.001. PMID 16243425. S2CID 22764367.
- ^ Doxakis E (April 2010). "Post-transcriptional regulation of alpha-synuclein expression by mir-7 and mir-153". начальный. Журнал биологической химии. 285 (17): 12726–34. Дои:10.1074/jbc.M109.086827. ЧВК 2857101. PMID 20106983.
- ^ Pieper HC, Evert BO, Kaut O, Riederer PF, Waha A, Wüllner U (December 2008). "Different methylation of the TNF-alpha promoter in cortex and substantia nigra: Implications for selective neuronal vulnerability". начальный. Нейробиология болезней. 32 (3): 521–7. Дои:10.1016/j.nbd.2008.09.010. PMID 18930140. S2CID 8673158.
- ^ Mogi M, Harada M, Narabayashi H, Inagaki H, Minami M, Nagatsu T (June 1996). "Interleukin (IL)-1 beta, IL-2, IL-4, IL-6 and transforming growth factor-alpha levels are elevated in ventricular cerebrospinal fluid in juvenile parkinsonism and Parkinson's disease". начальный. Письма о неврологии. 211 (1): 13–6. Дои:10.1016/0304-3940(96)12706-3. PMID 8809836. S2CID 54279479.
- ^ Bönsch D, Lenz B, Kornhuber J, Bleich S (February 2005). "DNA hypermethylation of the alpha synuclein promoter in patients with alcoholism". начальный. NeuroReport. 16 (2): 167–70. Дои:10.1097/00001756-200502080-00020. PMID 15671870. S2CID 43289612.
- ^ Jowaed A, Schmitt I, Kaut O, Wüllner U (May 2010). "Methylation regulates alpha-synuclein expression and is decreased in Parkinson's disease patients' brains". начальный. Журнал неврологии. 30 (18): 6355–9. Дои:10.1523/JNEUROSCI.6119-09.2010. ЧВК 6632710. PMID 20445061.
- ^ Desplats P, Spencer B, Coffee E, Patel P, Michael S, Patrick C, Adame A, Rockenstein E, Masliah E (March 2011). "Alpha-synuclein sequesters Dnmt1 from the nucleus: a novel mechanism for epigenetic alterations in Lewy body diseases". начальный. Журнал биологической химии. 286 (11): 9031–7. Дои:10.1074/jbc.C110.212589. ЧВК 3059002. PMID 21296890.
- ^ а б Outeiro TF, Kontopoulos E, Altmann SM, Kufareva I, Strathearn KE, Amore AM, Volk CB, Maxwell MM, Rochet JC, McLean PJ, Young AB, Abagyan R, Feany MB, Hyman BT, Kazantsev AG (July 2007). "Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson's disease". начальный. Наука. 317 (5837): 516–9. Bibcode:2007Sci...317..516O. Дои:10.1126/science.1143780. PMID 17588900. S2CID 84493360.
- ^ Song C, Kanthasamy A, Jin H, Anantharam V, Kanthasamy AG (October 2011). "Paraquat induces epigenetic changes by promoting histone acetylation in cell culture models of dopaminergic degeneration". начальный. Нейротоксикология. 32 (5): 586–95. Дои:10.1016/j.neuro.2011.05.018. ЧВК 3407036. PMID 21777615.
- ^ Harrison IF, Dexter DT (October 2013). "Epigenetic targeting of histone deacetylase: therapeutic potential in Parkinson's disease?". рассмотрение. Фармакология и терапия. 140 (1): 34–52. Дои:10.1016/j.pharmthera.2013.05.010. PMID 23711791.