Спиноцеребеллярная атаксия 1 типа - Spinocerebellar ataxia type 1
| Спиноцеребеллярная атаксия 1 типа | |
|---|---|
| Другие имена | SCA1, болезнь Шута |
 | |
| AXH домен Атаксин 1 | |
| Специальность | Неврология |
| Симптомы | Атаксия походки и стойки, гиперметрический саккады, дизартрия, дисфагия |
| Осложнения | пневмония, телесные повреждения в результате падений |
| Обычное начало | Между 3 и 4 декадами |
| Продолжительность | Долгосрочный |
| Причины | Генетический |
| Диагностический метод | Генетическое тестирование |
| Прогноз | 10–30 лет от начала |
| Частота | 1–2 на 100 000 |
Спиноцеребеллярная атаксия 1 типа (SCA1) - редкий аутосомно-доминантный расстройство, которое, как и другие спиноцеребеллярная атаксия, характеризуется неврологическими симптомами, включая дизартрия, гиперметрический саккады, и атаксия походки и стойки. Эта дисфункция мозжечка прогрессивный и постоянный. Первые симптомы обычно возникают в возрасте от 30 до 40 лет, хотя могут возникать и подростковые заболевания. Смерть обычно наступает в течение 10-30 лет от начала заболевания.
SCA1 обычно наследуется от родителей по аутосомно-доминантному типу; дети человека с этим заболеванием имеют 50% шанс унаследовать его сами, и в некоторых случаях могут возникать новые мутации. Это вызвано увеличением количества тринуклеотидные повторы в полиглутаминовый тракт из ATXN1 ген, кодирующий белок атаксин 1. Это расширение приводит к большему, чем обычно, количеству повторов нуклеотидной последовательности. цитозин, аденин, гуанин, или CAG в гене, что, в свою очередь, приводит к большему, чем обычно, количеству последовательных глутамин аминокислотные остатки в белке. Этот мутантный белок вызывает деградацию в определенных типах нейронов, таких как Нейроны Пуркинье, которые распространены в мозжечок, спинной мозг и связанные с ним части мозга. Хотя механизм до конца не изучен, предполагается, что изменения во взаимодействиях между атаксином 1 и другими белками приводят к токсическому усилению функции.
Мутация может быть обнаружена до или после появления симптомов с помощью генетическое тестирование. В настоящее время не известно лекарство от SCA1, поэтому лечение заболевания в первую очередь направлено на устранение симптомов для поддержания качество жизни, сфокусироваться на физиотерапия переобучить и заменить утраченные функции. Исследования по разработке методов лечения продолжаются, и в дополнение к традиционному фармацевтическому лечению SCA1 был предметом исследования более продвинутых вариантов лечения, таких как генная терапия и терапия стволовыми клетками. Во всем мире от 1 до 2 человек из 100 000 страдают спиноцеребеллярной атаксией 1 типа, однако распространенность варьируется между популяциями и часто связано с эффект учредителей.
Атаксия как симптом известна с середины 19 века, а гетерогенная группа заболеваний, ныне известная как спиноцеребеллярная атаксия, была предметом обширных исследований во второй половине того же века. Достижения в молекулярная генетика в 20 веке позволили выявить различные причины этих болезней. В начале 1990-х ген, вызывающий SCA1, был локализован в человеческий лейкоцитарный антиген комплекс на хромосома 6 и к 1993 году атаксин 1 был идентифицирован как ген, вызывающий заболевание. Это был первый ген, вызывающий спиноцеребеллярную атаксию, который был локализован и идентифицирован.
Признаки и симптомы
Атаксия относится к отсутствию скоординированных движений мышц, которые включают аномалия походки и это мозжечок признак, который типичен для всех типов спиноцеребеллярной атаксии (SCA), хотя у людей с SCA1 также развивается пирамидальный и бульбарный признаки по мере прогрессирования заболевания. Средний возраст дебюта - от 30 до 40 лет, хотя бывают исключения. От первых симптомов продолжительность обычно составляет от одного до трех десятилетий, где более раннее начало коррелирует с более быстрым прогрессированием.[1]
Спиноцеребеллярная атаксия 1, как и другие ВКА, часто вызывает дизартрия моторное расстройство речи часто проявляется невнятностью слов; патологический нистагм, расстройство, при котором глаза непроизвольно смещаются, что влияет на зрение; и проблемы с походкой и балансом. SCA1 также обычно присутствует с дисфагия, расстройство глотания, которое может вызвать удушье во время еды и питья; и гиперметрический саккады, где глаз имеет тенденцию двигаться быстрее или дальше, чем предполагалось, когда он отслеживает объект или перемещается от одного фокуса к другому. По мере прогрессирования заболевания могут появиться более серьезные неврологические симптомы, например: дисметрия, где движения конечностей постоянно превышают желаемое положение; дисдиадохокинезия, где повторяющиеся движения тела становятся нескоординированными; или же гипотония, где атрофируются мышцы. В то время как новые симптомы появляются по мере прогрессирования SCA1, нистагм может исчезнуть по мере замедления движений глаз и саккад. В конечном итоге летальный исход может быть вызван потерей бульбарных функций, но осложнениями из-за таких симптомов, как пневмония от проблем с глотанием, или травма от падений, также может быть смертельной.[1] Выраженность и точный фенотип этих симптомов могут варьироваться в зависимости от типа ВКА. Дизартрия SCA 1 может различаться по степени тяжести в зависимости от задачи и часто связана с более напряженной, задушенной или резкой вокализацией, чем при других расстройствах.[2]
Из-за значительных различий между случаями SCA1 типичные признаки и симптомы могут появляться вместе с более тонкими или редкими симптомами. Макулопатия сообщалось в редких случаях, и это может быть связано с эффектами мутации на ATXN1 локус на генах в соседних локусах.[3] Специфические дистонии сообщалось в отдельных случаях, часто в виде писательские судороги[4] или же шейная дистония.[5]
SCA также можно обнаружить до серьезной атрофии с электрофизиологический методы, использующие электроды на коже черепа для обнаружения изменений электрического потенциала в мозгу в ответ на ощущения или движения. У людей с SCA1 часто наблюдаются аномальные слуховой вызванный потенциал ствола мозга, включая длительную задержку и отсутствие или плохо определенные формы волны, при этом в одном исследовании сообщалось, что 73,3% испытуемых демонстрируют отклонения. В том же исследовании были обнаружены аномалии зрительный вызванный потенциал и средний соматосенсорный вызванный потенциал у некоторых SCA1 человек. Эти результаты были аналогичны результатам, полученным для других ВКА, а различия между ВКА не были статистически значимыми, поэтому электрофизиологические методы не могут заменить генетическое тестирование для конкретных диагнозов ВКА.[6]
Все SCA вызывают атрофию в различных нервных тканях, которые можно обнаружить с помощью магнитно-резонансная томография, компьютерная томография, или же другие методы визуализации. В SCA1 некоторая деградация в серое вещество мозжечка и ствола головного мозга иногда можно обнаружить у бессимптомных лиц с расширением в ATXN1.[7] Обычно потерю серого вещества можно наблюдать в червь мозжечка во всех дольках мозжечка и в парамедианных частях обоих полушарий. белое вещество потеря также может наблюдаться в середине ножки мозжечка. Потеря объема может быть связана с серьезностью и продолжительностью.[8]
По оценкам, в 77% случаев прогрессирующей болезни мозжечка имеется один или несколько расстройства психического здоровья, а 19% выставляют когнитивные расстройства.[9] Эти оценки постоянно выше, чем доля психических расстройств среди населения в целом, но все же следуют другим общим шаблонам, таким как корреляция между частотой депрессии и полом или возрастом. Неясно, может ли депрессия быть причинно связана с дегенерацией мозжечка; одно исследование сообщает о данных, согласующихся с тем, что депрессия в первую очередь является реакцией на инвалидность, а не ее симптомом,[10] в то время как другой сообщает о доказательствах того, что депрессия может иметь причинную связь; Распространенность депрессии у разных типов ВСА различается по-разному, чем скорость прогрессирования инвалидности.[11]
Генетика
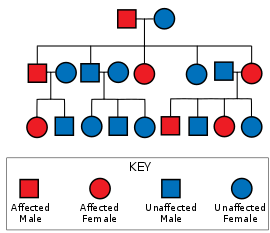
Спиноцеребеллярная атаксия 1 типа вызвана мутацией в ATXN1 ген. Эта мутация передается через аутосомно-доминантный паттерн наследования, означающий, что болезнь не пропускает поколения, по крайней мере один из родителей должен иметь болезнь, чтобы дети унаследовали ее, и что шансы того, что любой конкретный ребенок унаследует SCA 1, независимо от пола или других фенотипов, составляет 50%, если пострадавший родитель гетерозиготный.[12]:26 В ATXN1 ген на хромосома 6 кодирует белок атаксин 1, который используется в сигнальные пути и генная регуляция, и сильно выражается в Нейроны Пуркинье. Кодирующая область для Атаксина 1 (6p22.3[13]) содержит полиглутаминовый тракт переменной длины. SCA1 присутствует у людей, у которых область по крайней мере на одной копии хромосомы 6 содержит 39 или более непрерывных повторов глутамин в котором большее количество повторов коррелирует с более ранним началом и более быстрым прогрессированием. Гистидин перебои в работе полиглуатаминового тракта могут уменьшить или предотвратить SCA1.[14]
SCA1, как известно, проявляет генетическое ожидание, где у одного поколения с заболеванием может быть более раннее начало и более быстрое развитие, чем у предыдущего поколения. Это обычно вызвано расширением полиглутаминового тракта между поколениями и чаще встречается в случаях отцовского наследования. Этот неменделирующее наследование аналогично тому, что наблюдается в болезнь Хантингтона и считается, что это вызвано различиями в различных механизмах гамета продуктивность между полами, что приводит к увеличению мозаика в мужском зародышевый.[15] ДНК с повторами CAG склонна к образованию вторичных структур, в том числе петли для шпилек и R-петли, что может привести к мутациям и мозаицизму, если Ремонт ДНК механизмы выходят из строя. Эти вторичные структуры вызывают соматический мозаицизм из-за отставания ДНК-полимераза в Фрагменты Окадзаки и нарушая Ремонт несоответствия ДНК, базовая эксцизионная пластика, эксцизионная репарация нуклеотидов и механизмы восстановления двухцепочечных разрывов. Механизм расширения зародышевой линии не совсем понятен, но считается, что только пути репарации несоответствия влияют на нестабильность зародышевой линии и MSH2 белок репарации был связан с расширением мужских гамет на моделях мышей.[15]
Патофизиология
Нормальный атаксин 1 принимает непосредственное участие в ряде сигнальные пути, в белке убиквитинирование, Метаболизм РНК, в транскрипция регуляция, трансформация белков и стабилизация белков.[16][17]:149–165 Среди других взаимодействий он образует комплекс транскрипции с Связанный с ретиноидом фактор транскрипции орфанного ядерного рецептора α (RORα) после взаимодействия с активатором, Гистонацетилтрансфераза KAT5, иногда обозначаемый как TIP60,[18] и это в передаче сигналов, опосредованной метаботропный рецептор глутамата 1 (mGluR1).[19] Моделирование резонансного распознавания белка атаксина 1 показали возможные сайты связывания для независимый от фактора роста репрессор транскрипции 1 (Gfi-1). Прогнозы этой вычислительной модели показывают взаимодействие, которое может играть роль в патологии SCA1, поскольку известно, что белок Gfi-1 вызывает избирательную деградацию клеток Пуркинье.[17]:149–165 Именно активное участие атаксина 1 во многих различных функциях затрудняет идентификацию и понимание биохимической патофизиологии его мутантной формы.[17]:15
Механизм, с помощью которого расширенные области повторов CAG в атаксине 1 вызывают дегенерацию нейронов, неясен. Исторически считалось, что это вызвано агрегирование и отложение пораженного белка аналогично другим болезням распространения полиглутамина,[20] однако исследования на моделях грызунов показали значительно более позднее формирование ядерные включения мутантных белков в нейронах мозжечка и спинного мозга, чем в кортикальных нейронах и нейронах гиппокампа, которые обычно демонстрируют только легкую дегенерацию у лиц с SCA1, предполагая более сложный механизм.[21] Показано, что атаксин-нулевые мыши демонстрируют сниженное моторное и пространственное обучение, что позволяет предположить, что атаксин 1 играет роль в синаптической пластичности и взаимодействиях между мотонейронами и мышцами. гиппокамп. Однако у мышей, лишенных обеих копий атаксина 1, не развиваются прогрессирующие неврологические симптомы или признаки атрофии, что позволяет предположить, что токсичность мутированного белка, а не потеря функции, является основным механизмом патологии SCA1.[22] Сравнение мРНК мышей с нулевым атаксином и мышей с атаксином1154Q / + показывает, что есть общие изменения в экспрессии генов, включая активацию генов, которые, как известно, подавляются атаксином 1 /CIC сложный. Это указывает на то, что, хотя это и не является основным механизмом, потеря функции атаксина 1 вносит вклад в патогенез SCA1.[23] В то время как комплекс атаксин 1 / CIC теряет часть своей регуляторной функции с расширенным атаксином 1, у мышей с нокаутом CIC не наблюдается дегенерации, что позволяет предположить, что взаимодействия между атаксином 1 и CIC опосредуют большинство токсических эффектов.[24] Также известно, что мутантный атаксин-1 изменяет нейронную схему развивающегося мозжечка, что может привести к более поздней уязвимости клеток Пуркинье и предполагает существование не автономной клетки токсичности.[25]
Различные взаимодействия атаксина 1 приводят к множеству возможных факторов, которые могут усиливать или снижать токсичность его мутантной формы. Атаксин 1 дикого типа быстро разрушается в цитоплазме, но может быть стабилизирован фосфорилирование и 14-3-3 переплет по мере необходимости ячейке. SCA1-положительные мыши с гаплодефицитом в 14-3-3ε+/- было показано, что не наблюдается дегенерации мозжечка, но по-прежнему наблюдается летальная бульбарная дегенерация, что позволяет предположить, что атрофия мозжечка может быть связана с повышенной стабильностью расширенного белка атаксина 1 и что могут быть разные патогенные механизмы для разных областей мозга.[26] Сайт фосфорилирования - это серин у 776-го остатка в атаксине 1. Подобно мышам, у которых отсутствуют белки 14-3-3, мыши с этим остатком заменены на аланин не проявляют мозжечкового синдрома.[27] Точно так же удаление домена AXH из атаксина 1 предотвращает аберрантные взаимодействия с независимым от фактора роста репрессором транскрипции 1, что приводит к деградации GFI1 в протеасома. Расширение полиглутаминовой области приводит к увеличению сродства домена AXH атаксина 1 к определенным факторам транскрипции, и этот эффект, как полагают, играет значительную роль в токсичности атаксина 1.[28] Другой белок, который, как было показано, значительно взаимодействует с атаксином 1, - это богатый лейцином кислотный ядерный белок или LANP. Его функция неизвестна, но он преимущественно экспрессируется в тех же нейронах, что и атаксин 1, и, как было показано, локализуется в ядрах этих нейронов в тех же субструктурах, что и атаксин 1. LANP взаимодействует только с полиглутаминовой областью атаксина 1 и его взаимодействия тем сильнее, чем больше количество остатков глутамина, поэтому два белка, вероятно, жизненно важны для функций друг друга в нейронах, а LANP также может способствовать патологии мутантных белков атаксина 1.[29] Атаксин 1 нравится, также называемый Brother of Ataxin 1 или Boat, имеет значительные взаимодействия с атаксином-1 и многими ассоциированными белками, такими как N-CoR. Подобный атаксину 1 имеет пониженную экспрессию на моделях трансгенных мышей и, как было показано, снижает цитотоксичность атаксина-1.[30]
Токсичность мутировавшего белка вызывает деградацию нервных тканей. Это включает потерю дендритный прерывание или ветвление на ранних стадиях прогрессирования болезни и возможная атрофия тканей мозга на более поздних стадиях.[21] SCA1 вызывает умеренную деградацию различных тканей, включая оба полушария мозжечка, червь мозжечка, то мосты, а мозговой ствол. Это также вызывает легкую атрофию у мозговой кортикальный ткань.[31] Недавнее исследование также обнаружило значительную атрофию спинной мозг и сглаживание задний столбец и обнаружили корреляцию между площадью шнура, повторами CAG и оценками SARA в SCA1.[32] В тканях центральной нервной системы, в отличие от тканей костей, мышц или кожи, отсутствуют механизмы для эндогенной генерации и дифференциации новых клеток, а также для восстановления паттернов и связей на большом расстоянии по мере их утраты, поэтому по мере прогрессирования дегенерации потери являются постоянными.[33]
Диагностика и оценка
Большинство ВКА и других атаксических расстройств клинически неоднородны, что означает, что клинические признаки и симптомы схожи для разных заболеваний, и различить заболевания с помощью одного только неврологического обследования затруднительно.[1] У лиц с симптомами для диагностики заболеваний, связанных с атаксией, часто требуется неврологическое обследование, оценка состояния неврологический и история семьи, и молекулярные генетическое тестирование. Отсутствие семейного анамнеза не исключает наследственных причин, таких как спиноцеребеллярная атаксия 1 типа, поскольку семейный анамнез может не быть собран или может быть недоступен для определенных людей, а новые случаи могут возникать из-за ожидания аллеля с изменяемым числом повторов.[34]:2–4 Для постановки диагноза в настоящее время коммерчески доступно молекулярно-генетическое тестирование 14 типов SCA, включая SCA1. В тех случаях, когда SCA отсутствуют в семейном анамнезе или если семейный анамнез недоступен, тестирование на 4 наиболее распространенных SCA даст положительные результаты в 50% случаев с подозрением на SCA.[34]:11 Лица, которые подвержены риску наследования SCA1, но в настоящее время пребывают перед симптомами, также могут пройти молекулярно-генетическое тестирование.[35]
Генетическое тестирование
Генетическое тестирование - единственный окончательный способ дифференцировать типы спиноцеребеллярной атаксии из-за сходства клинических характеристик этих заболеваний и большой разницы между случаями. Генетическое тестирование доступно для многих типов SCA, включая относительно распространенные типы SCA1, 2, 3, 6, и 7; и менее распространенные SCA8, 10, 12, 14 и 17.[35] Однако генетическое тестирование стоит дорого и имеет низкую диагностическую ценность: положительные диагнозы обнаруживаются только в 24% тестов, заказанных узким специалистом, и в 10% в целом.[36]
Генетическое тестирование можно проводить на различных стадиях прогрессирования заболевания. Когда генетическое тестирование проводится после появления симптомов, тест считается диагностическим; у взрослых до появления симптомов он протекает бессимптомно, и тест может быть выполнен для пренатальной или доимплантационной диагностики. В Европейская сеть молекулярной генетики качества (EMQN) рекомендует критерии для каждого типа, которые должны быть соблюдены перед началом тестирования. EQMN рекомендует, чтобы лаборатории получали письменную клиническую оценку симптомов неврологом и раскрытие семейного анамнеза или его отсутствия до начала диагностического генетического тестирования.[37][38] Поскольку никаких профилактических или лечебных методов лечения SCA не известно, генетическое тестирование лиц из группы риска не рекомендуется для всех случаев и обычно проводится на индивидуальной основе.[39] Пресимптоматическое, дородовое и предимплантационное тестирование обычно запрашивается через генетический консультант и требовать наличия семейного анамнеза и документации об информированном согласии консультанта.[37][38] Спиноцеребеллярная атаксия 1-го типа была одним из первых заболеваний с поздним началом, в отношении которого пресимптоматическое тестирование оказалось эффективным и прогностическим; до разработки тестирования на SCA1 болезнь Хантингтона была единственным подобным заболеванием, для которого было доступно досимптоматическое тестирование.[40]
Молекулярно-генетическое тестирование SCA должно позволять дифференцировать образцы с патогенным аллелем от образцов без аллеля и уметь точно измерять количество повторов при нарушениях роста повторов. Капиллярный электрофорез (CE) - это один из методов, который отвечает этим критериям и рекомендован EMQN.[37][38] Другой распространенный метод - электрофорез в полиакриламидном геле (СТРАНИЦА). Оба метода требуют амплификации всех интересующих локусов для данного теста. Усиление осуществляется с помощью полимеразные цепные реакции или ПЦР. Выбор праймеров может позволить либо амплифицировать один ген, либо амплифицировать множество генов для использования в мультиплексный анализ что может сэкономить время в тех случаях, когда может потребоваться панель из множества тестов. И PAGE, и CE используют синхронизированные циклы электричества для протягивания кусочков ДНК через пористый полимер, разделяя аналиты по комбинации ионной подвижности, размера и массы. CE имеет преимущество перед PAGE в том, что измерения молекулярной массы, такие как масс-спектрометрии может использоваться с аналитами, тогда как PAGE требует использования Саузерн-блот чтобы позволить сравнение с лестница последовательности.[41] Для длин повторов в диапазоне, в котором важны прерывания, такие анализы, как CE и PAGE, не будут определять, является ли штамм патогенным, и потребуется дополнительное тестирование.[37][38]
Клинический
Для большинства ВКА не существует формальных диагностических критериев, и генетическое тестирование является единственным определенным диагностическим методом, но клиническое исследование признаков и симптомов может иметь жизненно важное значение для отличия ВКА от негенетических атаксий и от других типов генетических атаксий. Клиническое обследование также может в некоторой степени помочь различить типы SCA, поэтому генетические тесты для определенных типов могут иметь приоритет над другими. Диагностика SCA часто начинается с выявления симптомов, указывающих на нарушение мозжечка, таких как прогрессирующая атаксия или дизартрия, или с распознавания симптомов, аналогичных случаю, выявленному в семейном анамнезе, особенно у родственников первой или второй степени родства.[1] Многие лабораторные исследования могут быть использованы для дальнейшего сужения потенциальной причины атаксии; визуализация головного и спинного мозга и различные электрофизиологические исследования могут быть полезны для определения фенотипов заболевания, а исследования крови и мочи могут исключить приобретенные причины.[34]:4
При оценке атаксических расстройств и способов их лечения существует множество тесты, которые может выполнить невролог. Тесты могут оцениваться индивидуально или по шкале для оценки атаксии. Обследование мозжечка может включать в себя произнесение фраз с большим количеством согласных для обнаружения сканирование речи, обнаруживая горизонтальный взгляд нистагм следя за пальцем глазами, выполняя быстрые чередующиеся движения, например, многократно вращая рукой от ладони к спине, проверяя Феномен отскока Холмса, и тестирование коленный рефлекс при гипотонии или гипертонии.[42] Общие шкалы включают Международная кооперативная рейтинговая шкала атаксии (ICARS) и Шкала оценки и рейтинга атаксических расстройств (SARA) для оценки тяжести атаксии как симптома. ICARS измеряет по шкале 100, где 0 - нормальная функция, а 100 - максимально возможное нарушение, присваивая разные балльные значения для разных тестов.[43] Тесты разделены на категории, оценивающие осанку и походку, кинетические функции, речь и глазодвигательные функции. Хотя эти категории создают полезную категоризацию для оценки того, на каких областях необходимо сосредоточить внимание в терапии, эта избыточность приводит к увеличению времени тестирования, что может исказить результаты тестов, выполненных в конце сеанса; и может привести к противоречивым результатам.[44] SARA - более короткий экзамен, оцениваемый по шкале от 0 до 40, где снова ноль - нормальная функция, а 40 - максимально возможное нарушение. Он состоит из восьми тестов: походка, стойка, погоня за пальцами, тест от пальца к носу, быстрые чередующиеся движения рук, скольжение пятки и голени и три кинектических функциональных теста конечностей.[45]
Дифференциальная диагностика
Дифференциальная диагностика SCA клиническими методами затруднено, потому что эти заболевания клинически неоднородны и существует значительная разница между проявлением отдельных случаев. Использование клинической информации для дифференциальной диагностики используется для определения приоритета генетического тестирования, а не как самостоятельной диагностики. Было обнаружено много потенциальных дифференцирующих симптомов, и были разработаны методы оценки многих симптомов и их прогрессирования для руководства генетическим тестированием. Даже если не удается сразу определить конкретный тип спиноцеребеллярной атаксии история болезни, семейный анамнез, клиническое обследование могут помочь отличить другие атаксии и уменьшить количество генетических тестов, необходимых для определения типа ВКА. Обследование родственников людей со спорадической атаксией часто может выявить достаточно семейный анамнез, чтобы определить способ передачи.[46]
Есть несколько общих тенденций, которые могут быть полезны для различения SCA. SCA1 имеет тенденцию прогрессировать быстрее, чем SCA2, 3 и 6, с более значительным ежегодным изменением оценок SARA и более ранней потерей функций после начала.[47] В диагностике клинической атаксии визуализация может оказаться бесполезной для отличия SCA1 от других SCA, поскольку существует значительная разница между отдельными случаями и значительное совпадение между заболеваниями.[31] Вестибулоокулярный рефлекс можно протестировать, используя записанное видео импульсный тест головы или vHIT. В этом тесте SCA1 обычно имеет нормальную задержку рефлекса и не всегда демонстрирует дефицит функции VOR, что отличает его от SCA3 и атаксии Фридрейха.[48] Определенные паттерны глазных моторных расстройств, обнаруживаемые с помощью видеоокулография, похоже, типизируют определенные типы SCA. Хотя SCA1 существенно не коррелировал с уникальным паттерном, другие возможные SCA могут быть связаны, и отсутствие вертикального нистагма после горизонтального покачивания головой снижает вероятность диагноза SCA6, в то время как отсутствие прямоугольного волнового паттерна во время фиксация снижает вероятность SCA3.[49]
Одна из возможных систем дифференциальной диагностики типов SCA - это регистрация прогрессирования симптомов и использование Байесовская вероятность построить прогностическую модель или байесовский классификатор, который сравнивает наблюдаемые данные с тенденциями, подобными описанным выше, чтобы определить вероятность правильности каждого диагноза. Было показано, что один такой байесовский классификатор точно предсказывает 78% случаев SCA из когорты с известными типами SCA. В чувствительность и специфичность для SCA1 в этой модели было 76,9% и 98,2% соответственно. Региональные различия в распространенности, симптомах и клинической оценке могут по-прежнему ограничивать использование этой системы в больших масштабах, хотя система может внедряться отдельными клиниками с использованием их собственных региональных данных.[50]
Управление
В настоящее время нет лекарства от спиноцеребеллярной атаксии типа 1. Однако с некоторыми из ее симптомов можно справиться с помощью физический, профессиональный или же речь терапии, изменения образа жизни и диеты или с помощью лекарств. Устранение симптомов не предотвратит прогрессирование болезни, но может иметь важное значение для поддержания качество жизни.[12]:48 Однако важно отметить, что существует множество расстройств, вызывающих атаксию и связанные с ней симптомы, и что стратегии лечения, которые работают для некоторых, например: витамин Е добавки для лечения определенных приобретенных атаксий не будут работать при наследственных атаксиях, таких как SCA1, и могут быть опасны для здоровья человека.[12]:52
Небольшие когортные исследования показали, что люди с мозжечковыми расстройствами восстанавливают координацию и имеют более низкие баллы SARA независимо от стадии или тяжести их атаксии до терапии, когда они регулярно проходят физиотерапию или exergaming над людьми, которых нет. Эти исследования показывают, что многодоменная физиотерапия, более целенаправленная координационная тренировка и упражнения с интенсивными играми - все это привело к улучшению показателей SARA, эквивалентным по крайней мере одному году нормального прогресса, в среднем 2,2 балла или более, в течение нескольких недель. Хотя эти результаты являются многообещающими, для подтверждения этих результатов могут потребоваться более масштабные исследования.[51] В целом, физиотерапия для людей с атаксией имеет скромные доказательства, подтверждающие ее эффективность, но в современной практике используются индивидуальные методы лечения без стандартной процедуры принятия решений между клиниками, что ограничивает возможность воспроизводимой оценки качества рутинных процедур в литературе.[52] Среди наиболее ранних практик нейрореабилитации: Упражнения Френкеля, разработанный Генрихом Френкелем в середине девятнадцатого века;[53] эти упражнения были взяты из современных физиотерапия и реабилитация методы, называемые лечебной гимнастикой, и повседневные занятия, такие как вставание со стула, чтобы найти упражнения, которые тесно связаны с патологией атаксии и основаны на медленной практике и настойчивости людей. заново изучить ключевые моторные навыки, заменив потерянный проприоцепция с визуальной обратной связью. Существуют упражнения для нижних конечностей, такие как разгибание ног, и для верхних конечностей, например, установка колышков на доску, и в зависимости от степени атаксии их можно выполнять лежа, сидя или стоя. Все упражнения часто начинаются с простых движений, и им становится все труднее имитировать движения реального мира, затронутые заболеванием.[54]
Общие рекомендации для людей с дисфагией или проблемами глотания включают: пюре питание, замена в рационе трудно съедаемых продуктов или изменение позы во время еды. Когда проблемы с глотанием становятся настолько серьезными, что аспирационная пневмония становится частой или изменения в диете не могут предотвратить потерю веса, питательная трубка можно рассмотреть.[12]:82–86 Обычно это чрескожная эндоскопическая гастростомия трубки тонкой кишки (PEG-J), однако они не обязательно приводят к снижению частоты аспирации, поскольку засорение может привести к гастроэфагеальному рефлюксу, который может быть аспирирован. Прямые PEG-J, по-видимому, реже вызывают рефлюкс и имеют меньшую частоту аспирационной пневмонии по сравнению со стандартной процедурой PEG-J.[55] Были исследованы многочисленные стратегии лечения дисфагии, включая физические упражнения, такие как модифицированные Маневры Вальсальвы, фармацевтическое лечение было направлено на лечение спастичности и компенсирующие методы, включая корректировку осанки и более длительное жевание. Эти стратегии, как и лечение многих симптомов наследственной атаксии, имеют небольшие доказательства их полезности, но еще не установлены крупными исследованиями.[56]
Как и в случае со всеми наследственными заболеваниями, опасения по поводу воздействия на членов семьи, особенно на детей, часто очень важны. Лица с диагнозом SCA 1 могут искать генетическое консультирование чтобы помочь в планирование семьи, развивающиеся навыки совладания, и планирование на будущее. Лица с SCA 1 могут рассмотреть экстракорпоральное оплодотворение с доимплантационным тестированием, чтобы предотвратить передачу болезни детям.[35]
Прогноз
Проникновение для SCA1 составляет 100% для большинства аллелей, поэтому почти у всех людей, у которых есть хотя бы одна копия мутировавшего гена, в конечном итоге разовьются симптомы.[47] Сообщалось по крайней мере об одном случае, когда пенетрантность могла быть неполной у женщины с 44 глютаминовыми повторами с прерываниями гистидина, у отца которых были симптомы, но у нее не было симптомов в возрасте 66 лет.[1][57] Люди с низким числом повторов, от 39 до 55, обычно доживают до репродуктивного возраста и могут передать болезнь своим детям, в то время как большое количество повторов может указывать на ювенильное начало и летальный исход.[58]
Эпидемиология
Национальный институт здоровья сообщает, что SCA1 имеет распространенность примерно 1 или 2 на 100 000[59] однако обзор литературы показал, что эти оценки значительно различаются от исследования к исследованию и могут составлять менее 1 на 100 000 или доходить до 6 на 100 000.[60] Среди всех типов SCA1 является одним из наиболее распространенных, и доля, приходящаяся на SCA1, варьируется в зависимости от географического региона, при этом доля SCA1 достигает 40% от всех диагнозов SCA в популяциях в России и Южной Африке. В США SCA1 составляет 6% диагнозов SCA.[61] В целом на SCA1 приходится 6-27% всех случаев доминантной атаксии.[34]:6 Из-за позднего начала, часто появляющегося после репродуктивного возраста, SCA1 оказывает низкое интенсивность отбора, занимая около 0,19 места в рейтинге Индекс ворона, но интенсивность может меняться со временем в пределах популяции или семьи, поскольку ожидание увеличивает количество повторов CAG. Одним из следствий этого является то, что SCA1 вряд ли исчезнет из популяции одним лишь естественным отбором.[58]
Распространенность каждого типа SCA зависит от географического региона и этнической принадлежности, возможно, из-за эффекты основателя и исторические модели миграции.[62] Регионы с высокой распространенностью включают центральные Польша, где 68% аутосомно-доминантных мозжечковых расстройств - это SCA1;[63] сообщества в Тамил Наду, где до 7,2% населения имеет SCA1 в некоторых небольших селах;[64] то Регион Тохоку на северной части Остров Хонсю, при этом 24,8% случаев приходится на SCA1;[65] и среди Якутский населения в восточных Сибирь, с распространенностью 46 на 100 000 сельского населения.[58]
История
Атаксию как симптом впервые описал французский невролог. Дюшенн де Булонь в теме с tabes dorsalis.[66] К концу 19-го и началу 20-го веков обширные исследования характеристик, причин и диагностики наследственной мозжечковой атаксии проводились при работе нескольких выдающихся неврологов, в том числе Жан-Мартен Шарко, Пьер Мари, Николаус Фридрейх, Адольф Струмпелл, и другие. Мари описал ряд случаев наследственного заболевания, развившегося у взрослых, которое, по его мнению, клинически отличалось от Атаксия Фридрейха, спастическая параплегия, и другие известные типы атаксии, называемые синдромом наследственной мозжечковой атаксии, хотя она стала известна как атаксия Мари.[67]
Хотя наследственные паттерны были четко различны, в 1940-х годах продолжались дискуссии о том, действительно ли атаксия Мари отличается от атаксии Фрейдрейха и параплегии Штрюмпеля и представляют ли эти категории одно заболевание или несколько. Это произошло из-за гетерогенной природы наследственной атаксии, сходства симптомов и отсутствия понятных биохимических механизмов.[68] Дальнейшее разочарование по поводу двусмысленности терминов, введенных Мари и Фридрейхом, привело к созданию других систем классификации атаксий. Гордон Морган Холмс и Годвин Гринфилд каждая из них разработала системы классификации атаксий, что привело к появлению категорий, называемых оливопонтоцеребеллярной атрофией.[69] и спиноцеребеллярная деградация, хотя между системами был достигнут небольшой консенсус, и многие термины используются как синонимы.[66]
в эпоха депрессии США Семья Шут в Миннесоте была одной из семей, у которых была наследственная атаксия. Несколько членов семьи активно участвовали в исследовании, и семья согласилась на вскрытие мозга нескольких умерших родственников. Было обнаружено, что заболевание в семье Шута имеет аутосомно-доминантный тип наследования и поражает спиноцеребеллярный тракт. В 1945 г. Джон Шут получил бесплатное медицинское образование за службу в армии США во время Второй мировой войны и начал свои собственные усилия по исследованию наследственной атаксии.[70]:90–91 У Шута развилась атаксия, как и у многих его родственников. В 1957 году, когда атаксия Шута прогрессировала до такой степени, что он был не в состоянии продолжать обычную медицинскую практику, он основал Национальный фонд атаксии с лабораторным помещением, подаренным больницей Гленвуд-Хиллз в Миннеаполисе.[70]:131
Племянник Джона Шута, Лаверенс Шут, также стал исследователем атаксии и внес свой вклад в локализацию гена спиноцеребеллярной атаксии в человеческий лейкоцитарный антиген комплекс в хромосоме 6.[71] Успех связывания одного из этих классов болезней с локусом показал, что используемые системы классификации неспособны различать болезни с множеством различных причин. Многие атаксические расстройства, которые исторически идентифицировались как атаксия Мари, оливопонтоцеребеллярная атрофия или другие названия, теперь были переклассифицированы как типы спиноцеребеллярной атаксии, каждый тип пронумерован по мере обнаружения нового локуса.[72] В 1993 году были идентифицированы ген и мутация, вызывающие спиноцеребеллярную атаксию 1 типа. Это был первый известный генетический дефект, вызывающий атаксическое расстройство.[73]
Направления исследований
Особый интерес для исследователей представляет лечение и смягчение нейродегенеративных расстройств, и несколько потенциальных вариантов SCA1 изучаются. Поскольку патология SCA1 сложна, существует несколько возможных подходов к лечению, которые включают клиренс расширенных белков атаксина 1, снижение токсичности расширенных белков атаксина 1, подавление продукции атаксина 1, терапию множественными генами и замену потерянных клеток мозга.[74][75][16] Поскольку многие SCA, включая SCA1, являются полиглютаминовыми заболеваниями и действуют по механизмам, аналогичным механизмам, характерным для болезни Хантингтона, многие многообещающие методы лечения болезни Хантингтона также исследуются на предмет SCA.[62]
Подавление и молчание генов
Поскольку спиноцеребеллярная атаксия часто связана с мутацией одного гена, изменяя как ген выражается может изменить фенотип. Существует несколько подходов к изменению экспрессии мутантных белков, включая методы, полностью останавливающие экспрессию, известные как подавление гена. В SCA1 патогенез требует постоянной экспрессии мутанта. ATXN1 ген, и было показано, что молчание останавливает дальнейшее прогрессирование заболевания, очищает ядерные включения и агрегаты и приводит к частичному восстановлению двигательных функций на моделях грызунов с условной экспрессией гена. Условное выражение ATXN1 в моделях на мышах отличается от того, как ген был бы подавлен терапевтически, но результаты показывают, что терапевтические методы подавления гена могут быть жизнеспособными для лечения и управления SCA1.[76] Процесс, который превращает закодированную информацию в ДНК в белки, требует двух этапов: транскрипции, в которой ДНК используется для создания комплементарной цепи РНК с помощью РНК-полимеразы, и трансляции, в которой РНК используется для производства белка с помощью рибосом. Нарушение любого из этапов может замедлить или предотвратить экспрессию мутантного гена.
Атаксин 1 участвует в ряде сигнальных путей, и его экспрессия контролируется сигнальными путями. В MAPK / ERK было показано, что этот путь активирует экспрессию атаксина 1, и MSK1 также фосфорилирует атаксин 1, контролируя его локализацию и деградацию. Ингибиторы ключевых белков этого пути могут быть использованы в комбинированная терапия для потенциального снижения экспрессии и более низких концентраций устойчивого состояния атаксина 1.[77]
Один метод для прерывания перевода, антисмысловая олигонуклеотидная терапия, который использует одиночные цепи РНК, комплементарной мишени, чтобы предотвратить связывание мишени с рибосомой и вызвать деградацию мишени, уже начал клинические испытания при других нейродегенеративных заболеваниях с множеством различных механизмов доставки.[78] Похожая техника РНК-интерференция или РНКи. Вместо комплементарных «антисмысловых» цепей РНК, РНКи использует очень маленькие двухцепочечные сегменты РНК, называемые малая интерферирующая РНК что вызывает деградацию цели до того, как она может быть переведена. Исследования с использованием агентов РНКи, проведенные аденоассоциированные вирусы (AAV), как было показано, останавливает прогрессирование заболевания и приводит к некоторому восстановлению функции при лечении, применяемом только к глубоким ядрам мозжечка у мышей.[79] и макаки резус.[80] Оба этих метода трудно применить к полиглутаминовым заболеваниям, потому что нацеливание на полиглутаминовый тракт может также вызвать подавление регуляции нормальных генов. SCA1 также показал, что трудно надежно нацеливаться с однонуклеотидные полиморфизмы ограничение количества способов, которыми могут быть разработаны методы РНКи и антисмысловой терапии для лечения SCA1.[81]
Снижение токсичности и увеличение выживаемости клеток
Из-за многочисленных взаимодействий атаксина-1 с другими белками методы снижения токсичности мутантного белка атаксина-1 часто изменяют экспрессию родственных белков. Например, атаксин-1-подобный имеет много общих доменов с атаксином-1, а сверхэкспрессия атаксин-1-подобного конкурирует с атаксином-1 и предотвращает его интеграцию в другие комплексы, снижая токсичность.[82] Этот эффект был воспроизведен на моделях мышей с использованием AAV, и было показано, что он так же эффективен, как и методы РНКи, в замедлении прогрессирования симптомов.[83] Аналогичным образом препарат баклофен, который используется для уменьшения спастичность в лицах с рассеянный склероз и родственных заболеваний, действует как агонист рецепторы гамма-аминомасляной кислоты типа B (ГАМКBР). Этот путь перекрестные помехи с путем mGluR1, который взаимодействует с белком атаксина 1 и белками, ответственными за локализацию и деградацию атаксина 1, что позволяет предположить, что баклофен может быть эффективным средством лечения SCA 1.[84]
Молекулярные шапероны представляют собой введенные белки, которые могут взаимодействовать с мутантным белком, снижая токсичность с помощью различных механизмов. Исследования на моделях мышей и Дрозофила модели показали, что белки теплового шока 40 и 70 могут снизить токсичность расширенных белков атаксина 1 и замедлить прогрессирование SCA1.[16]
Хотя в настоящее время не существует известного метода исключительно стимулирования сокращений полиглутамина in vivo, методы, использующие программируемые нуклеазы показали некоторые надежды в том, что они вызывают эти изменения in vitro. Программируемые нуклеазы - это белки, которые могут разрывать цепи ДНК рядом с последовательностями, которые ученые могут определить перед использованием. Это включает в себя CRISPR / Cas9, который использует белок, содержащийся в бактериях, и направляющую цепь РНК, и нуклеазы цинковых пальцев, которые используют сконструированные белки со специальными повторяющимися ДНК-связывающими доменами, чтобы направлять присоединенную нуклеазу. В исследовании сообщается, что как нуклеазы CRISPR, так и нуклеазы цинковых пальцев, которые полагаются на двухцепочечные разрывы, вызывают сокращения и расширения почти с одинаковой частотой, в то время как CRISPR, использующий мутантный вариант Cas9, Cas9 D10A или Cas9 никаз, который вызывает только разрывы отдельных нитей, вызывает в основном сокращения.[85]
У мышей митохондриальный нарушения способствуют прогрессированию SCA1.[86] Заметные изменения в Клетка Пуркинье митохондриальные белки совпадают с симптоматической фазой заболевания. Клетки Пуркинье у мышей SCA1 также претерпевают возрастные изменения в морфологии митохондрий. Кроме того, клетки Пуркинье мышей SCA1 нарушили электронно-транспортные комплексы и уменьшился АТФаза Мероприятия. Опыт мышей SCA1 увеличился окислительный стресс и увеличился окислительное повреждение ДНК.[86] Митохондриальная направленная антиоксидант Было обнаружено, что MitoQ замедляет появление SCA1-сцепленного невропатологии например, отсутствие моторная координация. MitoQ также предотвращал вызванный окислительным стрессом Повреждение ДНК и потеря клеток Пуркинье.[86]
Терапия замещения клеток
Один из исследуемых вариантов лечения: терапия стволовыми клетками, который пытается заменить мертвые ткани путем трансплантации стволовые клетки в пораженную область и либо стимулируя их дифференцироваться в желаемые типы клеток, либо позволяя им стимулировать эндогенные регенеративные механизмы. Эти методы представляют интерес для исследователей как возможное лечение нейродегенеративных заболеваний, но в настоящее время имеют ограниченный успех в моделях на животных и в исследованиях культур клеток in vitro.[16] Способность привитых клеток интегрироваться в желаемую ткань и адаптироваться к уникальным патологиям различных нейродегенеративных расстройств может быть серьезным ограничением для разработки методов лечения на основе стволовых клеток. Кроме того, ткани мозга часто зависят от сложного и сложного расположения нейронов; области мозга, которые не требуют точности в этих паттернах для функционирования, например полосатое тело затронутый болезнь Паркинсона который использует паракринная передача сигналов, как правило, дают лучшие результаты при лечении стволовыми клетками, чем системы, требующие точности, такие как мозжечок и мост.[33] Терапия стволовыми клетками может быть особенно трудной для восполнения потери нейронов Пуркинье как незатронутой. гранулярные клетки может предотвратить достижение аксонов глубокие ядра мозжечка с которыми взаимодействуют клетки Пуркинье. Несмотря на эти трудности, привитые нервные клетки-предшественники было показано, что они жизнеспособны и успешно мигрируют в желаемое место на моделях трансгенных мышей SCA1 и мезенхимальные стволовые клетки было показано, что они уменьшают потерю дендритного ветвления у мышей SCA1.[87] Положительные результаты были обнаружены на моделях мышей с использованием стволовых клеток плода. нейроэктодерма и взрослые стволовые клетки из боковые желудочки и зубчатые извилины.[17]:177–188 Использование собранных стволовых клеток в терапии стволовыми клетками требует иммуносупрессия чтобы хозяин не отказывался от трансплантатов; создание индуцированные плюрипотентные стволовые клетки из собственных клеток хозяина может снизить этот риск, и были проведены некоторые испытания при других нейродегенеративных заболеваниях.[17]:177–188
Рекомендации
- ^ а б c d е Опал П., Ашизава Т. (1993). «Спиноцеребеллярная атаксия 1 типа». GeneReviews. Вашингтонский университет, Сиэтл. PMID 20301363. Получено 1 июля 2017.
- ^ Сидтис Дж. Дж., Ан Дж. С., Гомес С., Сидтис Д. (июль 2011 г.). «Речевые характеристики, связанные с тремя генотипами атаксии». Журнал коммуникативных расстройств. 44 (4): 478–92. Дои:10.1016 / j.jcomdis.2011.03.002. ЧВК 3159076. PMID 21592489.
- ^ Лебранчу П., Ле Меур Дж., Магот А., Дэвид А., Верни С., Вебер М., Милеа Д. (сентябрь 2013 г.). «Макулопатия и спиноцеребеллярная атаксия 1 типа: новая ассоциация?». Журнал нейроофтальмологии. 33 (3): 225–31. Дои:10.1097 / WNO.0b013e31828d4add. PMID 23584155. S2CID 23511772.
- ^ Ходжа Г.А., Шривастава А., Гуге В.В., Чаудри Н. (2016). «Писательский спазм при спиноцеребеллярной атаксии 1 типа». Журнал неврологии в сельской практике. 7 (4): 584–586. Дои:10.4103/0976-3147.186980. ЧВК 5006475. PMID 27695243.
- ^ Кикучи А., Такеда А., Сугено Н., Миура Е., Като К., Хасэгава Т. и др. (2016). «Метаболические изменения мозга при шейной дистонии со спиноцеребеллярной атаксией 1 типа после терапии ботулотоксином». Медицина внутренних органов. 55 (14): 1919–22. Дои:10.2169 / internalmedicine.55.5843. PMID 27432104.
- ^ Чандран В., Джунджхунвала К., Пурушоттам М., Джайн С., Пал П.К. (июль 2014 г.). «Мультимодальные вызванные потенциалы при спиноцеребеллярной атаксии 1, 2 и 3 типа». Летопись Индийской академии неврологии. 17 (3): 321–4. Дои:10.4103/0972-2327.138519. ЧВК 4162021. PMID 25221404.
- ^ Якоби Х., Ритц К., дю Монсель С.Т., Бауэр П., Мариотти С., Нанетти Л. и др. (Июль 2013). «Биологические и клинические характеристики лиц с риском спиноцеребеллярной атаксии типов 1, 2, 3 и 6 в продольном исследовании RISCA: анализ исходных данных». Ланцет. Неврология. 12 (7): 650–8. Дои:10.1016 / S1474-4422 (13) 70104-2. PMID 23707147. S2CID 28605950.
- ^ Джинестрони А., Делла Нейв Р., Тесса С., Джаннелли М., Де Грандис Д., Пласмати Р., Салви Ф., Пьячентини С., Маскальчи М. (август 2008 г.). «Структурное повреждение мозга при спиноцеребеллярной атаксии 1 типа: исследование VBM». Журнал неврологии. 255 (8): 1153–8. Дои:10.1007 / s00415-008-0860-4. PMID 18438695. S2CID 5642656.
- ^ Леруа И., О'Хирн Е., Марш Л., Ликетсос К. Г., Розенблатт А., Росс КА, Брандт Дж., Марголис Р. Л. (август 2002 г.). «Психопатология у пациентов с дегенеративными заболеваниями мозжечка: сравнение с болезнью Хантингтона». Американский журнал психиатрии. 159 (8): 1306–14. Дои:10.1176 / appi.ajp.159.8.1306. PMID 12153822.
- ^ Schmitz-Hübsch T, Coudert M, Tezenas du Montcel S, Giunti P, Labrum R, Dürr A, et al. (Апрель 2011 г.). «Коморбидность депрессии при спиноцеребеллярной атаксии». Двигательные расстройства. 26 (5): 870–6. Дои:10.1002 / mds.23698. PMID 21437988.
- ^ Lo RY, Figueroa KP, Pulst SM, Perlman S, Wilmot G, Gomez C, Schmahmann J, Paulson H, Shakkottai VG, Ying S, Zesiewicz T., Bushara K, Geschwind M, Xia G, Yu JT, Lee LE, Ashizawa T. , Subramony SH, Kuo SH (январь 2016 г.). «Депрессия и клиническое прогрессирование при спиноцеребеллярной атаксии». Паркинсонизм и связанные с ним расстройства. 22: 87–92. Дои:10.1016 / j.parkreldis.2015.11.021. ЧВК 4695274. PMID 26644294.
- ^ а б c d Нэнси М (2003). Жизнь с атаксией: справочник и ресурсы (2-е изд.). Миннеаполис: Национальный фонд атаксии. ISBN 978-0-943218-12-0. LCCN 2003111597.
- ^ "Отчет по символу ATXN1 | Комитет по номенклатуре генов HUGO". www.genenames.org. Комитет по номенклатуре генов HUGO. Получено 28 августа 2017.
- ^ Zühlke C, Dalski A, Hellenbroich Y, Bubel S, Schwinger E, Bürk K (2002). «Спиноцеребеллярная атаксия 1 типа (SCA1): исследования корреляции фенотип-генотип в промежуточных аллелях». Европейский журнал генетики человека. 10 (3): 204–9. Дои:10.1038 / sj.ejhg.5200788. PMID 11973625.
- ^ а б Краус-Перротта С., Лагалвар С. (22 ноября 2016 г.). «Экспансия, мозаицизм и прерывание: механизмы мутации CAG-повтора при спиноцеребеллярной атаксии 1 типа». Мозжечок и атаксия. 3 (20): 20. Дои:10.1186 / s40673-016-0058-у. ЧВК 5118900. PMID 27895927.
- ^ а б c d Кан С., Хонг С. (июнь 2009 г.). «Молекулярный патогенез заболевания спиноцеребеллярной атаксией 1 типа». Молекулы и клетки. 27 (6): 621–7. Дои:10.1007 / s10059-009-0095-у. PMID 19572115. S2CID 207047842.
- ^ а б c d е Sunghoi H, изд. (2012). Атаксия: причины, симптомы и лечение. Нью-Йорк: Nova Science Publishers. ISBN 978-1-61942-867-6. LCCN 2011946004.
- ^ Серра Х.Г., Дювик Л., Зу Т., Карлсон К., Стивенс С., Йоргенсен Н., Лисхольм А., Беррайт Е., Зогби Х.Й., Кларк Х. Б., Андресен Дж. М., Орр Х. Т. (ноябрь 2006 г.). «Опосредованное RORalpha развитие клеток Пуркинье определяет тяжесть заболевания у взрослых мышей SCA1». Клетка. 127 (4): 697–708. Дои:10.1016 / j.cell.2006.09.036. PMID 17110330.
- ^ Шуваев А.Н., Хосой Н., Сато Й., Янагихара Д., Хираи Х. (январь 2017 г.). «Прогрессирующее нарушение передачи сигналов mGluR мозжечка и его терапевтический потенциал для мозжечковой атаксии у мышей модели спиноцеребеллярной атаксии типа 1». Журнал физиологии. 595 (1): 141–164. Дои:10.1113 / JP272950. ЧВК 5199750. PMID 27440721.
- ^ Шастрый Б.С. (июль 2003 г.). «Нейродегенеративные нарушения агрегации белков». Neurochemistry International. 43 (1): 1–7. Дои:10.1016 / S0197-0186 (02) 00196-1. PMID 12605877. S2CID 31191916.
- ^ а б Ватасе К., Уибер Э.Дж., Сюй Б., Анталфи Б., Юва-Пейлор Л., Хашимото К., Кано М., Аткинсон Р., Сан И., Армстронг Д.Л., Сватт Д.Д., Орр Х.Т., Пейлор Р., Зогби Х.Ю. «Длинный повтор CAG в локусе Sca1 мыши воспроизводит особенности SCA1 и показывает влияние растворимости белка на селективную нейродегенерацию». Нейрон. 34 (6): 905–19. Дои:10.1016 / S0896-6273 (02) 00733-X. PMID 12086639. S2CID 18901202.
- ^ Каммингс CJ, Zoghbi HY (сентябрь 2000 г.). «Тринуклеотидные повторы: механизмы и патофизиология». Ежегодный обзор геномики и генетики человека. 1 (1): 281–328. Дои:10.1146 / annurev.genom.1.1.281. PMID 11701632.
- ^ Креспо-Баррето Дж., Фрайер Дж. Д., Шоу Калифорния, Орр Х. Т., Зогби Х.Й. (июль 2010 г.). «Частичная потеря функции атаксина-1 способствует нарушению регуляции транскрипции в патогенезе спиноцеребеллярной атаксии 1 типа». PLOS Genetics. 6 (7): e1001021. Дои:10.1371 / journal.pgen.1001021. ЧВК 2900305. PMID 20628574.
- ^ Rousseaux, Maxime W.C .; Чумперлин, Тайлер; Лу, Сян-Чжи; Lackey, Элизабет П .; Бондарь, Виталий В .; Ван, Ин-Уи; Тан, Цюмин; Адамски, Кэролайн Дж .; Фридрих, Джиллиан; Твароски, Кирк; Чен, Вейли; Толар, Якуб; Хенцлер, Кристина; Шарма, Аджай; Баич, Александар; Линь, Дао; Дювик, Лиза; Лю, Чжандун; Силлитоэ, Рой В .; Zoghbi, Huda Y .; Орр, Гарри Т. (21 марта 2018 г.). «Комплекс ATXN1-CIC является основным фактором патологии мозжечка при спиноцеребеллярной атаксии 1 типа за счет механизма усиления функции». Нейрон. 97 (6): 1235–1243.e5. Дои:10.1016 / j.neuron.2018.02.013. ISSN 0896-6273. ЧВК 6422678. PMID 29526553.
- ^ Эдамаканти, ЧР; Do, J; Дидонна, А; Мартина, М; Opal, P (13 марта 2018 г.). «Мутантный атаксин1 нарушает развитие мозжечка при спиноцеребеллярной атаксии 1 типа». Журнал клинических исследований. 128 (6): 2252–2265. Дои:10.1172 / JCI96765. ЧВК 5983343. PMID 29533923.
- ^ Кохияма М.Ф., Лагалвар С. (5 мая 2016 г.). «Механизмы стабилизации и деградации цитоплазматического атаксина-1». Журнал экспериментальной неврологии. 9 (Дополнение 2): 123–9. Дои:10.4137 / JEN.S25469. ЧВК 4859447. PMID 27168726.
- ^ Emamian ES, Kaytor MD, Duvick LA, Zu T, Tousey SK, Zoghbi HY, Clark HB, Orr HT (2003). «Серин 776 атаксина-1 имеет решающее значение для индуцированного полиглутамином заболевания у трансгенных мышей SCA1». Нейрон. 38 (3): 375–87. Дои:10.1016 / s0896-6273 (03) 00258-7. PMID 12741986. S2CID 16892848.
- ^ Цуда Х., Джафар-Неджад Х., Патель А.Дж., Сун Й., Чен Х.К., Роуз М.Ф., Венкен К.Дж., Ботас Дж., Орр Х.Т., Беллен Х.Дж., Зогби Х.Й. (август 2005 г.). «Домен AXH атаксина-1 опосредует нейродегенерацию через взаимодействие с белками Gfi-1 / Senseless». Клетка. 122 (4): 633–44. Дои:10.1016 / j.cell.2005.06.012. PMID 16122429. S2CID 16706329.
- ^ Матилла А., Коши Б.Т., Каммингс С.Дж., Исобе Т., Орр Х.Т., Зогби Х.Й. (октябрь 1997 г.). «Богатый лейцином кислый ядерный белок мозжечка взаимодействует с атаксином-1». Природа. 389 (6654): 974–8. Bibcode:1997Натура.389..974М. Дои:10.1038/40159. PMID 9353121. S2CID 4383708.
- ^ Mizutani A, Wang L, Rajan H, Vig PJ, Alaynick WA, Thaler JP, Tsai CC (сентябрь 2005 г.). «Бот, белок домена AXH, подавляет цитотоксичность мутантного атаксина-1». Журнал EMBO. 24 (18): 3339–51. Дои:10.1038 / sj.emboj.7600785. ЧВК 1224676. PMID 16121196.
- ^ а б Döhlinger S, Hauser TK, Borkert J, Luft AR, Schulz JB (2008). «Магнитно-резонансная томография при спиноцеребеллярной атаксии». Мозжечок. 7 (2): 204–14. Дои:10.1007 / s12311-008-0025-0. PMID 18418677. S2CID 20200822.
- ^ Martins CR, Martinez AR, de Rezende TJ, Branco LM, Pedroso JL, Barsottini OG, Lopes-Cendes I, França MC (август 2017 г.). «Повреждение спинного мозга при спиноцеребеллярной атаксии 1 типа». Мозжечок. 16 (4): 792–796. Дои:10.1007 / s12311-017-0854-9. PMID 28386793. S2CID 30480275.
- ^ а б Росси Ф., Каттанео Э. (май 2002 г.). «Мнение: терапия нервными стволовыми клетками при неврологических заболеваниях: мечты и реальность». Обзоры природы. Неврология. 3 (5): 401–9. Дои:10.1038 / №809. PMID 11988779. S2CID 28637104.
- ^ а б c d Перлман С.Л. (2016). Оценка и лечение атаксических расстройств: обзор для врачей. Миннеаполис: Национальный фонд атаксии. ISBN 978-0-943218-14-4. LCCN 2007923539.
- ^ а б c О'Салливан Смит C, Майкельсон SJ, Беннетт Р.Л., Берд Т.Д. «Спиноцеребеллярная атаксия: сделать осознанный выбор в отношении генетического тестирования» (PDF). Вашингтонский университет, нейрогенетика. Национальный институт исследований инвалидности и реабилитации. Получено 4 августа 2017.
- ^ Фогель Б.Л., Викри Б.Г., Уолтон-Ветцель Дж., Либер Э., Браунер С.Х. (август 2013 г.). «Использование генетического тестирования перед направлением к узкому специалисту по поводу мозжечковой атаксии». Генетическое тестирование и молекулярные биомаркеры. 17 (8): 588–94. Дои:10.1089 / gtmb.2013.0005. ЧВК 3732434. PMID 23725007.
- ^ а б c d Секейрос Дж., Мартиндейл Дж., Сенека С., Джунти П., Камяряйнен О, Вольпини В. и др. (Ноябрь 2010 г.). «Руководство EMQN Best Practice для молекулярно-генетического тестирования SCA». Европейский журнал генетики человека. 18 (11): 1173–6. Дои:10.1038 / ejhg.2010.8. ЧВК 2987475. PMID 20179742.
- ^ а б c d Секейрос Дж., Сенека С., Мартиндейл Дж. (Ноябрь 2010 г.). «Консенсус и разногласия в лучших практиках молекулярно-генетического тестирования спиноцеребеллярной атаксии». Европейский журнал генетики человека. 18 (11): 1188–95. Дои:10.1038 / ejhg.2010.10. ЧВК 2987480. PMID 20179748.
- ^ ван де Варренбург Б. П., ван Гален Дж., Бош С., Бургундер Дж. М., Дюрр А., Джунти П., Клокгайд Т., Мариотти С., Пандольфо М., Рисс О. (апрель 2014 г.). «Консенсус EFNS / ENS по диагностике и лечению хронической атаксии в зрелом возрасте». Европейский журнал неврологии. 21 (4): 552–62. Дои:10.1111 / ene.12341. PMID 24418350.
- ^ Шримптон А.Е., Дэвидсон Р., Макдональд Н., Брок DJ (1993). «Пресимптоматическое тестирование на аутосомно-доминантную спиноцеребеллярную атаксию типа 1». Журнал медицинской генетики. 30 (7): 616–7. Дои:10.1136 / jmg.30.7.616. ЧВК 1016469. PMID 8411042.
- ^ Доршнер МО, Барден Д., Стивенс К. (2002). «Диагностика пяти заболеваний спиноцеребеллярной атаксии с помощью мультиплексной амплификации и капиллярного электрофореза». Журнал молекулярной диагностики. 4 (2): 108–13. Дои:10.1016 / S1525-1578 (10) 60689-7. ЧВК 1906987. PMID 11986402.
- ^ "Осмотр мозжечка | Стэнфордская медицина 25 | Стэнфордская медицина". stanfordmedicine25.stanford.edu. Получено 1 сентября 2017.
- ^ Джонс МК, Комбс-Миллер С.А. (февраль 2016 г.). «Характеристики измерения и клиническая полезность Международной шкалы оценки атаксии у лиц с наследственной атаксией». Архивы физической медицины и реабилитации. 97 (2): 341–342. Дои:10.1016 / j.apmr.2015.04.002. Получено 31 августа 2017.
- ^ Schmitz-Hübsch T, Tezenas du Montcel S, Baliko L, Boesch S, Bonato S, Fancellu R и др. (Май 2006 г.). «Надежность и валидность Международной шкалы оценки совместной атаксии: исследование на 156 пациентах с спиноцеребеллярной атаксией». Двигательные расстройства. 21 (5): 699–704. Дои:10.1002 / mds.20781. PMID 16450347.
- ^ Schmitz-Hübsch T, du Montcel ST, Baliko L, Berciano J, Boesch S, Depondt C и др. (Июнь 2006 г.). «Шкала оценки атаксии: разработка новой клинической шкалы». Неврология. 66 (11): 1717–20. Дои:10.1212 / 01.wnl.0000219042.60538.92. PMID 16769946. S2CID 24069559.
- ^ Дегардин А., Доббелэр Д., Вийом I, Дефоорт-Дельлеммес С., Хуртевент Дж. Ф., Саблоньер Б., Десте А., Дефевр Л., Девос Д. (март 2012 г.). «Спиноцеребеллярная атаксия: рациональный подход к этиологической диагностике». Мозжечок. 11 (1): 289–99. Дои:10.1007 / s12311-011-0310-1. PMID 21892625. S2CID 15108793.
- ^ а б Ашизава Т., Фигероа К.П., Перлман С.Л., Гомес К.М., Уилмот Г.Р., Шмахманн Д.Д., Ин Ш., Зесевич Т.А., Полсон Х.Л., Шаккоттай В.Г., Бушара К.О., Куо С.Х., Гешвинд М.Д., Ся Джи, Маццони П., Кришер Дж. , Holbert AR, Ferguson JH, Pulst SM, Subramony SH (ноябрь 2013 г.). «Клинические характеристики пациентов со спиноцеребеллярной атаксией 1, 2, 3 и 6 в США; проспективное обсервационное исследование». Журнал редких заболеваний Orphanet. 8 (1): 177. Дои:10.1186/1750-1172-8-177. ЧВК 3843578. PMID 24225362.
- ^ Луис Л., Коста Дж., Муньос Е., де Карвалью М., Кармона С., Шнайдер Е., Гордон С. Р., Вальс-Соле Дж. (Июль 2016 г.). «Вестибулоокулярная рефлекторная динамика с головными импульсами различает спиноцеребеллярную атаксию 1, 2 и 3 типов и атаксию Фридрейха». Журнал вестибулярных исследований. 26 (3): 327–34. Дои:10.3233 / VES-160579. PMID 27392837.
- ^ Ким Дж. С., Ким Дж. С., Юн Дж., Со Д. В., Чон Й, Кан Дж. Х., Пак Дж. Х., Чо Дж. У. (август 2013 г.). «Глазно-моторные характеристики различных подтипов спиноцеребеллярной атаксии: отличительные особенности». Двигательные расстройства. 28 (9): 1271–7. Дои:10.1002 / mds.25464. PMID 23609488.
- ^ Maschke M, Oehlert G, Xie TD, Perlman S, Subramony SH, Kumar N, Ptacek LJ, Gomez CM (ноябрь 2005 г.). «Профиль клинических признаков спиноцеребеллярной атаксии типа 1-8 предсказывает генетически определенные подтипы». Двигательные расстройства. 20 (11): 1405–12. Дои:10.1002 / mds.20533. PMID 16037936.
- ^ Синофзик М, Ильг В (2014). «Моторная тренировка при дегенеративном спиноцеребеллярном заболевании: улучшение атаксии с помощью интенсивной физиотерапии и игр с упражнениями». BioMed Research International. 2014: 583507. Дои:10.1155/2014/583507. ЧВК 4022207. PMID 24877117.
- ^ Мартин К.Л., Тан Д., Брагге П., Бялоцерковский А. (январь 2009 г.). «Эффективность физиотерапии для взрослых с дисфункцией мозжечка: систематический обзор». Клиническая реабилитация. 23 (1): 15–26. Дои:10.1177/0269215508097853. PMID 19114434. S2CID 25458915.
- ^ Zwecker M, Zeilig G, Ohry A (январь 2004 г.). «Профессор Генрих Себастьян Френкель: забытый основатель реабилитационной медицины». Спинной мозг. 42 (1): 55–6. Дои:10.1038 / sj.sc.3101515. PMID 14713947.
- ^ Барклай, Х. В. (март 1913 г.). «Лечебная гимнастика при локомоторной атаксии: упражнения Френкеля и другие». Американский журнал медсестер. 13 (6): 428–436. Дои:10.2307/3403902. JSTOR 3403902.
- ^ Панайотакис PH, ДиСарио Дж.А., Хильден К., Огара М., Фанг Дж.С. (апрель 2008 г.). «Установка трубки DPEJ предотвращает аспирационную пневмонию у пациентов из группы высокого риска». Питание в клинической практике. 23 (2): 172–5. Дои:10.1177/0884533608314537. PMID 18390785.
- ^ Фогель А.П., Кейдж М.Дж., Йоханссон К., Шаллинг Э. (ноябрь 2015 г.). «Лечение дисфагии (затруднения глотания) при наследственной атаксии». Кокрановская база данных систематических обзоров (11): CD010169. Дои:10.1002 / 14651858.cd010169.pub2. PMID 26564018.
- ^ Гольдфарб Л.Г., Васконселос О., Платонов Ф.А., Лункес А., Кипнис В., Кононова С., Чабрашвили Т., Владимирцев В.А., Алексеев В.П., Гайдусек Д.К. (апрель 1996 г.). «Нестабильный триплетный повтор и фенотипическая изменчивость спиноцеребеллярной атаксии 1 типа». Анналы неврологии. 39 (4): 500–6. Дои:10.1002 / ana.410390412. PMID 8619528. S2CID 38240020.
- ^ а б c Платонов Ф.А., Тырышкин К., Тихонов Д.Г., Неустроева Т.С., Сивцева Т.М., Яковлева Н.В., Николаев В.П., Сидорова О.Г., Кононова С.К., Гольдфарб Л.Г., Ренвик Н.М. (июль 2016 г.). «Генетическая приспособленность и интенсивность отбора в популяции, пораженной высокочастотной спиноцеребеллярной атаксией 1 типа». Нейрогенетика. 17 (3): 179–85. Дои:10.1007 / s10048-016-0481-5. ЧВК 5262524. PMID 27106293.
- ^ «SCA1». Справочник по генетике. Национальный институт здоровья. Получено 19 июля 2017.
- ^ Руано Л., Мело С., Сильва М.С., Коутиньо П. (2014). «Глобальная эпидемиология наследственной атаксии и спастической параплегии: систематический обзор исследований распространенности». Нейроэпидемиология. 42 (3): 174–83. Дои:10.1159/000358801. PMID 24603320.
- ^ "Спиноцеребеллярная атаксия типа 1 | Центр атаксии | Чикагский университет". ataxia.uchicago.edu. Центр Атаксии Чикагского университета. Получено 13 июля 2017.
- ^ а б Soong BW (август 2004 г.). «Наследственные спиноцеребеллярные атаксии: количество, распространенность и перспективы лечения» (PDF). Гонконгский медицинский журнал = Xianggang Yi Xue Za Zhi. 10 (4): 229–30. PMID 15299166.
- ^ Крыса В., Сулек А., Ракович М., Ширковец В., Заремба Дж. (Август 2016 г.). «Высокая относительная частота SCA1 в Польше отражает потенциальный эффект основателя». Неврологические науки. 37 (8): 1319–25. Дои:10.1007 / с10072-016-2594-х. ЧВК 4956719. PMID 27193757.
- ^ Ренгарадж Р., Дханарадж М., Арулможи Т., Чаттопадхьяй Б., Баттачарья Н.П. (2005). «Высокая распространенность спиноцеребеллярной атаксии 1 типа в этнической тамильской общине в Индии». Неврология Индия. 53 (3): 308–10, обсуждение 311. Дои:10.4103/0028-3886.16929. PMID 16230798. S2CID 37292868.
- ^ Онодера Ю., Аоки М., Цуда Т., Като Х., Нагата Т., Камея Т., Абе К., Итояма Ю. (2000). «Высокая распространенность спиноцеребеллярной атаксии типа 1 (SCA1) в изолированном регионе Японии». Журнал неврологических наук. 178 (2): 153–8. Дои:10.1016 / S0022-510X (00) 00390-7. PMID 11018707. S2CID 34519174.
- ^ а б Клокгайд Т., Полсон Х (май 2011 г.). «Вехи в развитии атаксии». Двигательные расстройства. 26 (6): 1134–41. Дои:10.1002 / mds.23559. ЧВК 3105349. PMID 21626557.
- ^ Алмейда GM, Герминиани FM, Тейве HA (октябрь 2015 г.). «Основополагающая роль Пьера Мари в неврологии и внутренней медицине». Arquivos de Neuro-Psiquiatria (на португальском). 73 (10): 887–9. Дои:10.1590 / 0004-282X20150126. PMID 26331384.
- ^ Миндлин Х.С., Мелараньо Филхо Р. (июнь 1943 г.). "Считайте себя собственными как наследственные вырождения espinho-cerebelares: A proposito de dois casos de heredo-ataxia cerebelar de Pièrre Marie em duas irmãs". Arquivos de Neuro-Psiquiatria (на португальском). 1 (1): 26–42. Дои:10.1590 / S0004-282X1943000100004.
- ^ Холмс Г. (1908). «Попытка классифицировать болезнь мозжечка с примечанием о наследственной мозжечковой атаксии Мари». Мозг. 30 (4): 545–567. Дои:10.1093 / мозг / 30.4.545.
- ^ а б Schut HJ (1978). Десять лет жизни. Гранд-Рапидс, Мичиган: Компания Baker Book House. ISBN 978-0-8010-8127-9.
- ^ Моллема Н., Орр Х (2013). «Поиск одной семьи, чтобы объяснить фатальное неврологическое расстройство». Американский ученый. 101 (6): 442. Дои:10.1511/2013.105.442.
- ^ Кеппен, Арнульф Х. (1 июня 1998 г.). "Наследственные атаксии". Журнал невропатологии и экспериментальной неврологии. 57 (6): 531–543. Дои:10.1097/00005072-199806000-00001. PMID 9630233.
- ^ Орр Х.Т., Чанг М.Ю., Банфи С., Квятковски Т.Дж., Сервадио А., Боде А.Л., МакКолл А.Е., Дювик Л.А., Ранум Л.П., Зогби Х.Й. (июль 1993 г.). «Экспансия нестабильного тринуклеотидного повтора CAG при спиноцеребеллярной атаксии 1 типа». Природа Генетика. 4 (3): 221–6. Дои:10.1038 / ng0793-221. PMID 8358429. S2CID 8877695.
- ^ Pérez Ortiz, J.M .; Орр, Х. Т. (2018). «Спиноцеребеллярная атаксия 1 типа: молекулярные механизмы нейродегенерации и доклинические исследования». Достижения экспериментальной медицины и биологии. 1049: 135–145. Дои:10.1007/978-3-319-71779-1_6. ISBN 978-3-319-71778-4. PMID 29427101.
- ^ Buijsen, Ronald A.M .; Toonen, Lodewijk J.A .; Гардинер, Сара Л .; Ван Рун-Мам, Виллек М.С. (2019). «Генетика, механизмы и терапевтический прогресс при полиглутаминовой спиноцеребеллярной атаксии». Нейротерапия. 16 (2): 263–286. Дои:10.1007 / s13311-018-00696-у. ЧВК 6554265. PMID 30607747.
- ^ Zu T, Duvick LA, Kaytor MD, Berlinger MS, Zoghbi HY, Clark HB, Orr HT (октябрь 2004 г.). «Восстановление от индуцированной полиглутамином нейродегенерации у условных трансгенных мышей SCA1». Журнал неврологии. 24 (40): 8853–61. Дои:10.1523 / JNEUROSCI.2978-04.2004. ЧВК 6729947. PMID 15470152.
- ^ Пак Дж., Аль-Рамахи I, Тан Кью, Моллема Н., Диас-Гарсия Дж. Р., Гальего-Флорес Т., Лу Х. С., Лагалвар С., Дувик Л., Кан Х., Ли Й., Джафар-Нежад П., Сайег Л. С., Ричман Р., Лю X, Гао Y, Шоу CA, Артур Дж. С., Орр Х. Т., Уэстбрук Т. Ф., Ботас Дж., Зогби Х.Й. (июнь 2013 г.). «Путь RAS-MAPK-MSK1 модулирует уровни белка атаксина 1 и токсичность в SCA1». Природа. 498 (7454): 325–331. Bibcode:2013Натура.498..325П. Дои:10.1038 / природа12204. ЧВК 4020154. PMID 23719381.
- ^ Эверс М.М., Тоонен Л.Дж., ван Рун-Мам В.М. (июнь 2015 г.). «Антисмысловые олигонуклеотиды в терапии нейродегенеративных расстройств». Расширенные обзоры доставки лекарств. 87: 90–103. Дои:10.1016 / j.addr.2015.03.008. PMID 25797014.
- ^ Кейзер М.С., Будро Р.Л., Дэвидсон Б.Л. (март 2014 г.). «Широкий терапевтический эффект после доставки вектора экспрессии РНКи в глубокие ядра мозжечка: значение для терапии спиноцеребеллярной атаксии 1 типа». Молекулярная терапия. 22 (3): 588–595. Дои:10,1038 / мт.2013.279. ЧВК 3944323. PMID 24419082.
- ^ Кейзер М.С., Кордовер Дж. Х., Гонсалес-Алегре П., Дэвидсон Б.Л. (декабрь 2015 г.). «Широкое распространение сайленсинга атаксина 1 в резус-мозжечке для терапии спиноцеребеллярной атаксии типа 1». Мозг. 138 (Pt 12): 3555–66. Дои:10.1093 / мозг / awv292. ЧВК 4840549. PMID 26490326.
- ^ Фисер А., Олейничак М., Свитонски П.М., Вроблевска Дж. П., Вишневска-Крук Дж., Миковска А., Кшизосяк В. Дж. (Март 2012 г.). «Оценка терапевтических стратегий на основе олигонуклеотидов для лечения заболеваний polyQ». BMC Молекулярная биология. 13 (1): 6. Дои:10.1186/1471-2199-13-6. ЧВК 3359213. PMID 22397573.
- ^ Боумен А.Б., Лам Ю.К., Джафар-Неджад П., Чен Х.К., Ричман Р., Самако Р.С., Фрайер Д.Д., Кале Дж.Дж., Орр Х.Т., Зогби Х.Й. (март 2007 г.). «Дупликация Atxn1l подавляет невропатологию SCA1 за счет уменьшения включения атаксина-1, расширенного полиглутамином, в нативные комплексы». Природа Генетика. 39 (3): 373–9. Дои:10,1038 / ng1977. PMID 17322884. S2CID 37879597.
- ^ Кейзер М.С., Геогеган Дж.С., Будро Р.Л., Леннокс К.А., Дэвидсон Б.Л. (август 2013 г.). «РНКи или сверхэкспрессия: альтернативные методы лечения спиноцеребеллярной атаксии 1 типа». Нейробиология болезней. 56: 6–13. Дои:10.1016 / j.nbd.2013.04.003. ЧВК 4173078. PMID 23583610.
- ^ Ади В., Ватт А.Дж. (январь 2017 г.). «Новые старые препараты от спиноцеребеллярной атаксии». Журнал физиологии. 595 (1): 5–6. Дои:10.1113 / JP273149. ЧВК 5199732. PMID 28035676.
- ^ Cinesi C, Aeschbach L, Yang B, Dion V (ноябрь 2016 г.). «Сокращение повторов CAG / CTG с использованием никазы CRISPR-Cas9». Nature Communications. 7: 13272. Bibcode:2016НатКо ... 713272C. Дои:10.1038 / ncomms13272. ЧВК 5105158. PMID 27827362.
- ^ а б c Штуки Д.М., Рюгсеггер С., Штайнер С., Радеке Дж., Мерфи М.П., Зубер Б., Саксена С. (август 2016 г.). «Митохондриальные нарушения способствуют прогрессированию спиноцеребеллярной атаксии 1 типа и могут быть улучшены с помощью нацеленного на митохондрии антиоксиданта MitoQ». Свободный Радич. Биол. Med. 97: 427–440. Дои:10.1016 / j.freeradbiomed.2016.07.005. PMID 27394174.
- ^ Cendelin J (февраль 2016 г.). «Трансплантация и терапия стволовыми клетками при дегенерации мозжечка». Мозжечок. 15 (1): 48–50. Дои:10.1007 / s12311-015-0697-1. PMID 26155762. S2CID 13470632.
| Классификация | |
|---|---|
| Внешние ресурсы |