Лице-лопаточно-плечевая мышечная дистрофия - Facioscapulohumeral muscular dystrophy
| Лице-лопаточно-плечевая мышечная дистрофия | |
|---|---|
| Другие имена | Мышечная дистрофия Ландузи-Дежерина, ФСГМД, ФСГ |
| Интервал времени экспрессии DUX4 в мышечных клетках FSHD[1] | |
| Произношение | |
| Специальность | Неврология, нервно-мышечная медицина |
| Симптомы | Слабость лица, крыло лопатки, отвисание стопы |
| Обычное начало | Подростковый возраст |
| Продолжительность | Долгосрочный |
| Типы | FSHD1, FSHD2 |
| Причины | Генетический (унаследованная или новая мутация) |
| Диагностический метод | Генетическое тестирование |
| Дифференциальная диагностика | Мышечная дистрофия конечностей (особенно кальпаинопатия ), Болезнь Помпе, митохондриальная миопатия, полимиозит[2] |
| Управление | Физиотерапия, фиксация, реконструктивная хирургия |
| Частота | От 1 из 8333 до 1 из 15000[2] |

Лице-лопаточно-плечевая мышечная дистрофия (FSHD) является разновидностью мышечная дистрофия что преимущественно ослабляет скелетные мышцы из лицо (Латинский: фацио), те, которые занимают позицию в лопатка (скапуло), и те, что в верхней части руки, лежащий над плечевая кость кость (плечевой).[2] Слабость лопатных мышц приводит к неправильному расположению лопатки (крылатая лопатка ). Слабость обычно проявляется и в других частях тела, например в животе и голени. падение ноги. Две стороны тела часто поражаются неодинаково. Симптомы обычно начинаются в раннем детстве и становятся заметными в подростковом возрасте, при этом 95% заболевших проявляют болезнь к 20 годам.[3] Немышечные проявления ЛЛПД включают: потеря слуха и аномалии кровеносных сосудов в задняя часть глаза.
ЛЛПД вызывается сложными генетическими изменениями, связанными с DUX4 ген.[4] У тех, у кого нет FSHD, DUX4 выражается (то есть: включается) в раннем развитии человека, а затем подавляется (то есть: выключается) в зрелых тканях.[5] В FSHD, DUX4 неадекватно выключен, что может быть вызвано несколькими различными мутациями, наиболее распространенной из которых является делеция ДНК в области, окружающей DUX4.[6] Эта мутация называется "D4Z4 сокращение "и определяет ЛЛПД типа 1 (ЛЛПД1), составляющих 95% случаев ЛЛПД. ЛЛПД из-за других мутаций классифицируется как ЛЛПД типа 2 (ЛЛПД2). Независимо от того, какая мутация присутствует, болезнь может возникнуть только в том случае, если у человека есть 4qA аллель, который является распространенной вариацией в ДНК рядом с DUX4.[7] До 30% случаев ЛЛПД связаны с новой мутацией, которая затем может передаваться детям.[8] FSHD1 следует за аутосомно-доминантный закономерность наследования, означающая, что каждый ребенок затронутого человека имеет 50% -ный шанс быть затронутым.[2] Как DUX4 выражение вызывает повреждение мышц неясно.[2] Выражение DUX4 Ген производит белок DUX4, функция которого заключается в модуляции сотен других генов, многие из которых участвуют в работе мышц.[2][4] Диагноз ставится генетическое тестирование.[2]
Нет известного лекарства от ЛЛПД. Никакие фармацевтические препараты не доказали свою эффективность в изменении течения болезни. Симптомы можно лечить с помощью физиотерапии, фиксации и реконструктивной хирургии. Хирургическая фиксация лопатки к грудной клетке в отдельных случаях эффективна для уменьшения симптомов плечевого сустава.[9] FSHD - третий по распространенности генетическое заболевание скелетных мышц (Дюшенн /Мышечная дистрофия Беккера быть первым и миотоническая дистрофия будучи вторым), затрагивая от 1 из 8 333 до 1 из 15 000 человек.[2] Прогноз чрезвычайно разнообразен, многие из них никогда не сталкиваются со значительными ограничениями, хотя до 20% пострадавших людей становятся тяжелыми инвалидами, требующими использования инвалидная коляска или самокат для передвижения.[3] Продолжительность жизни обычно не поражается, за исключением редких случаев дыхательная недостаточность.[10]
Первое описание человека с ЛЛПД - это отчет о вскрытии трупа 1852 г.[11][12] хотя ЛЛПД не считалась болезнью до 1870-х и 1880-х годов, когда французские врачи Ландузи и Дежерин следил за семьей, пострадавшей от этого; поэтому FSHD иногда называют Мышечная дистрофия Ландузи-Дежерина.[13][12] В 1991 году была установлена связь большинства случаев с верхушкой хромосомы 4, что, как было обнаружено, связано с сокращением D4Z4 в 1993 году. DUX4 был открыт в 1999 году, но только в 2010 году генетический механизм, вызывающий его экспрессию, был выяснен. В 2012 году была обнаружена преобладающая мутация FSHD2. В 2014 году исследователи опубликовали первое предложенное патофизиологическое определение болезни и четыре жизнеспособные терапевтические цели для возможных точек вмешательства.[14]
Признаки и симптомы


Мышцы лица, плечевой пояс, и плечо обычно поражаются, хотя эти мышцы можно сохранить, а другие мышцы обычно поражаются. Распределение и степень мышечной слабости чрезвычайно различаются даже между однояйцевыми близнецами.[15][16] Отдельные мышцы могут ослабнуть, в то время как соседние мышцы остаются здоровыми.[нужна цитата ] Слабость мышц обычно становится заметной на одной стороне тела раньше другой, что является признаком болезни.[нужна цитата ] Мышцы правого плеча поражаются чаще, чем мышцы левого плеча, независимо от руки.[17]:139[18] Скелетно-мышечная боль очень распространена, чаще всего описывается в шее, плечах, пояснице и задней части колена.[19] Как правило, симптомы появляются в возрасте 15–30 лет, хотя также встречаются младенческое начало, начало у взрослого и отсутствие симптомов, несмотря на наличие причинной генетики.[8] Длительные статические фазы, в которых не наблюдается прогрессирования, не редкость.[20] FSHD1 и FSHD2 имеют схожие признаки и симптомы, хотя очень большие делеции D4Z4 в FSHD1 (ЭкоRI 10-11 кб ) сильнее связаны с младенческим началом, прогрессирующей потерей слуха, заболеваниями сетчатки и различными редкими проявлениями.[21]
Лицо и плечо
Слабость обычно начинается с мышц лица.[20] По крайней мере, легкая слабость лица может быть обнаружена у 90% и более пациентов с ЛЛПД, хотя это редко является первоначальной жалобой.[22] Мышцы, окружающие глаза (orbicularis oculi мышца ), что может привести к сну с открытыми веками.[нужна цитата ] Мышца, окружающая рот (orbicularis oris мышца ) также обычно поражается, что приводит к неспособности сморщить губы или свистеть.[нужна цитата ] Могут быть трудности с произнесением букв M, B и P или выражением лица, которое выглядит подавленным, подавленным, злым или усталым.[нужна цитата ] После слабости лица обычно развивается слабость в мышцах верхней части туловища, особенно в мышцах, соединяющих плечевой пояс с грудной клеткой. Слабость мышц плечевого пояса является первичной жалобой в 80% случаев, а в 30% семейных случаев болезнь не прогрессирует.[22] Преимущественно передняя зубчатая мышца и средние и нижние волокна трапеции под действием; верхние трапециевидные волокна часто сохраняются.[нужна цитата ]. Эта слабость приводит к тому, что лопатки становятся повернутый вниз и затяжной, в результате чего крылатые лопатки, горизонтальные ключицы и наклонные плечи. В запущенных случаях кажется, что лопатка «грыжаются» вверх и над грудной клеткой. Распространенная жалоба - трудности при работе с руками над головой. В вращающая манжета мышцы обычно сохраняются даже на поздних стадиях болезни.[23][24] Еще одна часто поражаемая мышца верхней части туловища - это большая грудная мышца, особенно стерно реберный участок, атрофия которого может способствовать выраженному горизонтальному передняя подмышечная складка.[25][8]
Верхняя часть руки и нижняя часть тела
После слабости лица и верхней части туловища слабость может «спуститься» на плечи (двуглавая мышца и трехглавая мышца ) и тазовый пояс.[20] Предплечья обычно сохраняются, в результате чего внешний вид можно сравнить с вымышленным персонажем. Попай.[8] Иногда наблюдается слабость, которая «пропускает» таз и вовлекает передняя большеберцовая мышца (мышца голени), вызывая падение ноги. Слабость также может возникать в мышцах живота, что может проявляться выпуклостью живота, поясничной гиперфункцией.лордоз, неспособность приседать или неспособность повернуться с одной стороны на другую в положении лежа. Нижние волокна прямая мышца живота поражаются чаще, чем верхние волокна, что проявляется как положительный Признак Бивора.[8] Слабость в ногах может проявляться затруднением ходьбы или легким сгибанием бедер.
Поражение мышц с точки зрения медицинской визуализации
Медицинская визуализация (КТ и МРТ) показала повреждение мышц, которое не вызывает явных симптомов.[23] Одно исследование МРТ показывает большая круглая мышца быть часто затронутым.[24] В полуперепончатая мышца, часть подколенные сухожилия, обычно поражается,[18][26][27] один автор заявил, что это «наиболее часто и серьезно поражаемая мышца».[2] Также МРТ показывает, что прямая мышца бедра поражается чаще, чем другие мышцы четырехглавой мышцы,[26] медиальный икроножная мышца поражается чаще, чем латеральная икроножная мышца,[26][27] и подвздошно-поясничная мышцы очень часто щадят.[27][2]
Немощно-скелетный
Наиболее частым немощно-скелетным проявлением ЛЛПД являются легкие аномалии кровеносных сосудов сетчатки, такие как телеангиэктазии или микроаневризмы, при этом в одном исследовании заболеваемость составила 50%.[нужна цитата ] Эти аномальные кровеносные сосуды обычно не влияют на зрение или здоровье, хотя их тяжелая форма имитирует Болезнь Пальто, состояние, обнаруживаемое примерно в 1% случаев ЛЛПД и чаще связанное с большими делециями 4q35.[2][28] Высокочастотная потеря слуха может возникать у людей с большими делециями 4q35, но в остальном встречается не так часто по сравнению с населением в целом.[2] Может быть нарушено дыхание, связанное с кифосколиоз и использование инвалидных колясок; это наблюдается у одной трети пациентов, прикованных к инвалидной коляске.[нужна цитата ] Однако поддержка аппарата искусственной вентиляции легких (ночная или дневная) требуется только в 1% случаев.[2][29]
Генетика
Генетика ЛЛПД сложна, что приводит к аномальным выражение из DUX4 ген.[2][6] У тех, у кого нет FSHD, DUX4 выражается во время эмбриогенез и в какой-то момент становится подавленный во всех тканях, кроме яички. При ЛЛПД наблюдается неадекватное подавление DUX4, позволяя эктопическое производство белка DUX4 в мышцах, вызывая повреждение мышц. Два генетических элемента необходимы для неадекватного подавления DUX4. Во-первых, должна быть мутация, вызывающая гипометилирование из ДНК окружающий DUX4, позволяя транскрипция из DUX4 в информационная РНК (мРНК). Несколько мутаций вызывают гипометилирование, в результате чего ЛЛД подразделяется на ЛЛД типа 1 (FSHD1) и FSHD типа 2 (FSHD2).[14]
Второй необходимый генетический элемент - это полиаденилирование последовательность вниз по течению к DUX4 что обеспечивает стабильность DUX4 мРНК, что позволяет DUX4 мРНК сохраняться достаточно долго, чтобы транслироваться в DUX4 белок, причинный агент повреждения мышц.[6] Есть как минимум 17 вариаций, или гаплотип полиморфизмы 4q35 (ДНК, включающие массив повторов D4Z4), наблюдаемые в популяции.[30] Эти 17 вариантов можно условно разделить на группы 4qA и 4qB.[30] Именно аллели 4qA содержат сигналы полиаденилирования, обеспечивающие стабильность DUX4 мРНК.[6] Аллели 4qB не имеют последовательностей полиаденилирования.[6]
DUX4 и массив повторений D4Z4
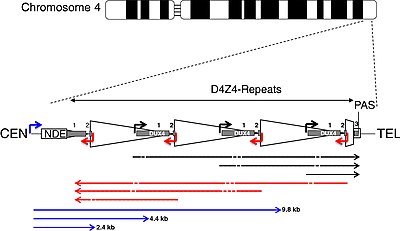
| CEN | центромерный конец | ТЕЛ | теломерный конец |
| Коробка NDE | неудаленный элемент | PAS | полиаденилирование сайт |
| треугольник | D4Z4 повтор | трапеция | частичный повтор D4Z4 |
| белая коробка | pLAM | серые коробки | DUX4 экзоны 1, 2, 3 |
| стрелки | |||
| угол | промоутеры | прямой | Транскрипты РНК |
| чернить | смысл | красный | антисмысловой |
| синий | DBE-T | тире | сайты по игре в кости |
DUX4 находится внутри массива макросателлитных повторов D4Z4, серии тандемно повторяющихся сегментов ДНК в субтеломерный область (4q35) из хромосома 4. Каждый повтор D4Z4 составляет 3,3 пары килобаз (kb) длинный и является участком эпигенетической регуляции, содержащим оба гетерохроматин и эухроматин конструкции.[31][32] При ЛЛД структура гетерохроматина теряется, превращаясь в эухроматин.[31] Название «D4Z4» происходит от устаревшей системы номенклатуры, используемой для сегментов ДНК неизвестного значения во время проект генома человека: D для ДНК, 4 для хромосомы 4, Z указывает, что это повторяющаяся последовательность, и 4 - серийный номер, присваиваемый в соответствии с порядком подачи.[33][34]
DUX4 состоит из трех экзонов. Экзоны 1 и 2 находятся в каждом повторе. Экзон 3 находится в области pLAM, теломерной по отношению к последнему частичному повтору.[6][5] Множественные транскрипты РНК продуцируются из массива повторов D4Z4, как смысловых, так и антисмысловых. Некоторые транскрипты могут деградировать на участках с образованием si-подобных малых РНК.[14] Некоторые транскрипты, которые берут начало в центромерных массивах повторов D4Z4 в неотделенном элементе (NDE), называемые транскриптами регуляторного элемента D4Z4 (DBE-T), могут играть роль в DUX4 дерепрессия.[14][35] Один из предложенных механизмов заключается в том, что DBE-T приводит к привлечению Белок группы триторакс Ash1L, увеличение H3K36me2 -метилирование и, в конечном итоге, дерепрессия генов 4q35.[36]
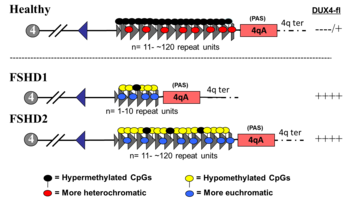
FSHD1
FSHD, включающий делецию повторов D4Z4 (так называемое «сокращение D4Z4»), классифицируется как FSHD1, что составляет 95% случаев FSHD.[2] Обычно хромосома 4 включает от 11 до 150 повторов D4Z4.[31][6] В FSHD1 имеется 1–10 повторений D4Z4.[6] Количество повторов примерно обратно коррелирует с тяжестью заболевания. А именно, те, у кого есть 1-3 повтора, с большей вероятностью будут иметь тяжелое, атипичное и раннее начало заболевания; те, у кого 4-7 повторов, имеют умеренное заболевание, которое сильно варьирует; а те, у кого есть 8-10 повторов, как правило, имеют самые легкие проявления, иногда без симптомов.[37] Сокращение D4Z4 вызывает гипометилирование D4Z4, позволяя DUX4 транскрипция. Удаление всего массива повторов D4Z4 не приводит к FSHD, потому что тогда нет полных копий DUX4 должны быть выражены, хотя в результате возникают другие врожденные дефекты.[38][6] Наследование аутосомно-доминантный, хотя от 10 до 30% случаев de novo (новый) мутации.[8]
Субтеломерная область хромосомы 10q содержит структуру тандемных повторов в высокой степени. гомологичный (99% идентично) 4q35.[6][30] Повторы 10q называются «D4Z4-подобными» повторами.[6] Поскольку в 10q обычно отсутствует последовательность полиаденилирования, он, как правило, не вовлечен в заболевание, за исключением случая хромосомные перестройки между 4q и 10q, приводящим к сокращению 4q D4Z4, или другому случаю переноса повтора 4q D4Z4 и сигнала полиаденилирования на 10q.[39][6]
FSHD2
ЛЛПД без сокращения D4Z4 классифицируется как ЛЛД2, что составляет 5% случаев ЛЛД.[2] Различные мутации вызывают FSHD2, все приводящие к гипометилированию D4Z4, при котором механизм заболевания сходится с FSHD1.[40] Около 80% случаев ЛЛД2 связаны с деактивирующими мутациями в гене. СМЧД1 (Структурное поддержание гибкого шарнирного домена хромосом, содержащего 1) на хромосома 18, ген, ответственный за метилирование ДНК.[2] СМЧД1 дезактивация приводит к гипометилированию массива повторов D4Z4. Другая причина FSHD2 - мутация в DNMT3B (ДНК-метилтрансфераза 3B), которая также играет роль в метилировании ДНК.[41][42] По состоянию на 2020 год ранние данные показывают, что третьей причиной FSHD2 является мутация в обеих копиях LRIF1 ген, кодирующий белок лиганд-зависимый фактор взаимодействия ядерных рецепторов 1 (LRIF1).[43] Известно, что LRIF1 взаимодействует с белком SMCHD1.[43] По состоянию на 2019 год, предположительно, существуют дополнительные мутации в других неопознанных генетических местах, которые могут вызвать FSHD2.[2]
Мутация одного аллеля СМЧД1 или же DNMT3B может вызвать болезнь. Мутация обеих копий LRIF1 Предварительно было показано, что к 2020 году он вызывает заболевание у одного человека.[43] Как и в случае FSHD1, для возникновения заболевания должен присутствовать аллель 4qA. Однако, в отличие от массива D4Z4, гены, участвующие в FSHD2, не находятся в непосредственной близости с аллелем 4qA, и поэтому они унаследован независимо от аллеля 4qA, что приводит к дигенному типу наследования. Например, один родитель без FSHD может передать СМЧД1 мутации, и другой родитель, также без ЛПД, может передать аллель 4qA, родив ребенка с ЛЛД2.[40][42]
Два конца спектра болезней
Первоначально FSHD1 и FSHD2 были описаны как две отдельные генетические причины одного и того же заболевания. Однако их также можно рассматривать не как отдельные причины, а как факторы риска. Нередко оба фактора способствуют развитию болезни у одного и того же человека.[37]
У пациентов с FSHD2, хотя у них нет аллеля 4qA с числом повторов D4Z4 меньше 11, они все же часто имеют один меньше 17 (относительно короткий по сравнению с общей популяцией), предполагая, что большое количество повторов D4Z4 может предотвратить эффекты СМЧД1 мутация.[37] Необходимы дальнейшие исследования, чтобы определить верхний предел повторов D4Z4, в которых может встречаться FSHD2.[37]
У тех, у кого аллель 4qA и 10 или меньше повторов, дополнительный СМЧД1 мутация усугубляет болезнь, классифицируя их как FSHD1 и FSHD2.[44] У этих людей с FSHD1 / FSHD2 паттерн метилирования массива повторов D4Z4 похож на наблюдаемый у FSHD2.[37] Такое комбинированное представление FSHD1 / FSHD2 наиболее часто встречается у пациентов с 9-10 повторами и редко встречается у пациентов с 8 или менее повторами. Относительное обилие СМЧД1 мутации в группе из 9-10 повторов вероятны, потому что значительная часть общей популяции имеет 9-10 повторов без заболевания, но с аддитивным эффектом СМЧД1 мутации, развиваются симптомы и ставится диагноз. У пациентов с 8 или менее повторами симптомы более вероятны, чем у пациентов с 9-10 повторами, что приводит к диагнозу независимо от дополнительных СМЧД1 мутация.[37]
Кажущаяся частота случаев FSHD1 / FSHD2 в диапазоне 9-10 повторов в сочетании с FSHD2-подобным паттерном метилирования указывает на то, что размер 9-10 повторов является зоной перекрытия между FSHD1 и FSDH2.[37]
Патофизиология

По состоянию на 2020 год, похоже, существует консенсус в отношении того, что ошибочное выражение DUX4 в мышцах является причиной ЛЛПД.[45] DUX4 выражается в очень малых количествах, обнаруживаемых в 1 из 1000 незрелые мышечные клетки (миобласты), который, по-видимому, увеличивается после созревания миобластов, отчасти потому, что клетки сливаются по мере созревания, и один ядро выражая DUX4 может обеспечить DUX4 белка в соседние ядра из слитых клеток.[46]
Остается областью активных исследований, как DUX4 вызывает повреждение мышц. DUX4 белок - фактор транскрипции, который регулирует многие другие гены. Некоторые из этих генов участвуют в апоптоз, Такие как p53, стр.21, МОЙ С, и β-катенин. Кажется, что DUX4 делает мышечные клетки более склонными к апоптозу, хотя детали механизма до сих пор неизвестны и оспариваются. Другой DUX4 регулируемые гены участвуют в окислительный стресс, и кажется, что DUX4 экспрессия снижает толерантность мышечных клеток к окислительному стрессу. Вариация способности отдельных мышц справляться с окислительным стрессом может частично объяснять паттерны вовлечения мышц при ЛЛПД. DUX4 подавляет многие гены, участвующие в развитии мышц, в том числе MyoD, миогенин, десмин, и PAX7. DUX4 показал сокращение мышечная клетка пролиферация, дифференциация и слияние. Эстроген кажется, играет роль в изменении DUX4 влияние на дифференцировку мышц, что может объяснить, почему женщины страдают меньше, чем мужчины. DUX4 регулирует несколько генов, которые участвуют в РНК контроль качества и DUX4 Экспрессия вызывает накопление РНК с последующим апоптозом.[45]
Сотовый гипоксия ответ, как сообщалось в одном исследовании, был основным драйвером DUX4-индуцированная гибель мышечных клеток. В факторы, вызываемые гипоксией (HIF) регулируются DUX4, возможно, вызывая патологические сигналы, ведущие к гибели клеток.[47]
Другое исследование показало, что DUX4 экспрессия в мышечных клетках привела к привлечению и изменению волокнистый /толстый клетки-предшественники, которые помогают объяснить, почему мышцы заменяются жиром и фиброзная ткань.[46]
Диагностика
Генетическое тестирование
Генетическое тестирование - это Золотой стандарт для диагностики ЛЛПД, так как это наиболее чувствительный и специфический тест доступен.[2] Обычно сначала тестируется FSHD1.[2] Укороченная длина массива D4Z4 (ЭкоRI длиной от 10 до 38 т.п.н.) со смежным аллелем 4qA поддерживает FSHD1.[2] Если FSHD1 отсутствует, обычно FSHD2 проверяется на предмет следующего, оценивая метилирование в 4q35.[2] Низкое метилирование (менее 20%) в контексте аллеля 4qA достаточно для диагностики.[2] Специфическая мутация, обычно одна из различных мутаций SMCHD1, может быть идентифицирована с помощью секвенирование следующего поколения (NGS).[48]
Оценка длины D4Z4
Измерение длины D4Z4 является технически сложной задачей из-за повторяющегося массива D4Z4, состоящего из длинных повторяющихся элементов.[49] Например, NGS бесполезен для оценки длины D4Z4, потому что он разбивает ДНК на фрагменты перед их считыванием, и неясно, из какого повтора D4Z4 произошел каждый секвенированный фрагмент.[8] В 2020 г. оптическое отображение стал доступен для измерения длины массива D4Z4, который является более точным и менее трудоемким, чем саузерн-блот.[50] Молекулярное расчесывание также доступен для оценки длины массива D4Z4.[51] Иногда 4q или 10q будут иметь комбинацию D4Z4 и D4Z4-подобных повторов из-за обмена ДНК между 4q и 10q, что может привести к ошибочным результатам, требующим более детальной проработки.[30]

Полиморфизма длин рестрикционных фрагментов Анализ (ПДРФ) был первым разработанным генетическим тестом, который до сих пор используется с 2020 года, хотя постепенно отменяется новыми методами. Это включает в себя разрезание ДНК на рестрикционные ферменты и сортировка полученных рестрикционные фрагменты по размеру с использованием Саузерн-блот. Рестрикционные ферменты ЭкоRI и МлрдЯ обычно используется. ЭкоRI изолирует массивы повторов 4q и 10q, и МлрдЯ делю последовательность 10q на мелкие кусочки, чтобы можно было различить 4q.[8][30] В ЭкоРестрикционный фрагмент RI состоит из трех частей: 1) проксимальной части 5,7 т.п.н., 2) центрального массива повторов D4Z4 переменного размера и 3) дистальной части, обычно 1,25 т.п.н.[52] Проксимальный участок имеет последовательность ДНК, окрашиваемую зондом p13E-11, который обычно используется для визуализации фрагмента EcoRI во время саузерн-блоттинга.[30] Название «p13E-11» отражает то, что это субклон последовательности ДНК, обозначенной как космид 13E во время проекта генома человека.[53][54] Иногда делеции массива повторов D4Z4 могут включать сайт связывания p13E-11, что требует использования альтернативных зондов.[30] Учитывая, что каждый повтор D4Z4 составляет 3,3 т.п.н., а длина ЭкоФрагмент RI содержит 6,9 т.п.н. ДНК, которая не является частью массива повторов D4Z4, количество единиц D4Z4 может быть рассчитано.
- D4Z4 повторы = (ЭкоДлина РИ - 6,9) / 3,3
Альтернативное тестирование

Когда стоимость непомерно высока или диагноз ЛЛПД не подозревается в качестве причины симптомов, пациенты и врачи могут полагаться на один или несколько из следующих тестов, все из которых менее чувствительны и менее специфичны, чем генетическое тестирование.[55]
- Креатинкиназа (CK) уровень в крови часто назначается при подозрении на повреждение мышц. СК является фермент обнаруживается в мышцах и попадает в кровь при повреждении мышц. Однако уровни КК при ЛЛПД повышены незначительно или даже в норме.[2]
- Электромиограмма (ЭМГ) измеряет электрическую активность мышцы. ЭМГ показывает неспецифические признаки мышечного повреждения или раздражительности.[2]
- Скорость нервной проводимости (NCV) измеряет, насколько быстро сигналы проходят от одной части нерва к другой. Нервные сигналы измеряются с помощью поверхностных электродов (аналогичных тем, которые используются для электрокардиограммы) или игольчатых электродов.
- Биопсия мышц включает хирургическое удаление небольшого фрагмента мышцы, обычно с руки или ноги. Биопсия оценивается с помощью различных биохимический тесты. Биопсия мышц, пораженных ЛЛД, показывает неспецифические признаки, такие как наличие лейкоцитов и изменение размера мышечных волокон. Этот тест назначается редко.[2]
- МРТ мышц позволяет обнаружить повреждение мышц даже в малосимптомных случаях. Из-за особых паттернов поражения мышц при ЛЛПД МРТ может помочь дифференцировать ЛЛПД от других мышечных заболеваний, направляя молекулярную диагностику.[23][24]
Управление
По состоянию на 2020 год не существует лекарства от ЛЛПД, и никакие фармацевтические препараты не доказали свою эффективность для изменения течения болезни. Упражнение аэробики было показано, что он снижает хроническую усталость и замедляет жировую инфильтрацию мышц при ЛЛПД.[56][57] Американская академия неврологии (ANN) рекомендует людям с ЛЛПД заниматься аэробными упражнениями низкой интенсивности для повышения уровня энергии, здоровья мышц и здоровья костей.[2] Силовые тренировки средней интенсивности не приносят вреда, хотя не доказано, что они приносят пользу.[58] Физиотерапия может устранить определенные симптомы; не существует стандартизованного протокола для FSHD. Отдельные сообщения предполагают, что при правильном применении кинезиологическая лента может уменьшить боль.[59] Трудотерапия можно использовать для обучения ежедневные занятия (ADL) и помочь адаптироваться к новым вспомогательные устройства. Когнитивно-поведенческая терапия (КПТ) снижает хроническую усталость при ЛЛПД, а также замедляет жировую инфильтрацию мышц, когда направлено на увеличение повседневной активности.[56][57]
Подтяжки часто используются для устранения мышечной слабости. Укрепление лопатки может улучшить положение лопатки, что улучшает функцию плеча, хотя часто считается неэффективным или непрактичным.[60] Ортезы голеностопного сустава может улучшить ходьбу, равновесие и качество жизни.[61]
Для выявления осложнений можно провести несколько медицинских тестов. Расширенный проверка зрения искать аномалии сетчатки рекомендуется тем, у кого впервые диагностирована ЛЛПД. Людей с большими делециями D4Z4 следует направлять к специалисту по сетчатке для ежегодных осмотров.[62][2] Проверка слуха должна проводиться у людей с ранним началом ЛЛПД до поступления в школу или у любого другого человека с ЛЛПД с симптомами потери слуха.[62][2] Исследование легочной функции (PFT) следует проводить у тех, кому впервые поставлен диагноз, для определения исходной легочной функции.[2] ПТФ также следует проводить периодически тем, у кого есть риск или симптомы легочной недостаточности.[62][2]
Хирургическое вмешательство
Различные проявления слабости лица поддаются хирургической коррекции. Золотые имплантаты верхнего века использовались для тех, кто не мог закрыть глаза.[63] Обвисшая нижняя губа была устранена с помощью пластической хирургии.[64] Некоторые случаи провисания стопы можно исправить хирургическим путем с помощью пересадки сухожилия, например, с помощью процедуры уздечки.[65][66][59] Причины тяжелого сколиоза, вызванного ЛЛПД, можно исправить с помощью спондилодеза.
Несколько процедур могут лечить лопаточное крыло, самая известная из которых - слияние лопаточно-грудного отдела (артродез ), ортопедическая процедура, при которой достигается сращение кости между лопаткой и ребрами. Увеличивает активность плеч диапазон движения, улучшает функцию плеч, уменьшает боль и улучшает внешний вид.[67][68] Активный диапазон движений больше всего увеличивается в условиях тяжелого крыла лопатки при неизмененном дельтовидная мышца;[9] однако диапазон пассивных движений уменьшается. А именно, пациент приобретает способность медленно сгибать и отводить плечи на 90+ градусов, но теряет способность «поднимать» руку на полные 180 градусов.[2] Второй тип процедуры - скапулопексия, которая включает привязку лопатки к ребрам, позвонкам или другим лопаткам с помощью трансплантатов сухожилий, проволоки или других средств. В отличие от лопаточно-грудного сращения, сращения между костями не происходит. Существует несколько типов скапулопексии, и результаты у каждого разные. По сравнению с лопаточно-грудными сращениями, скапулопексия считается менее инвазивной, но также более подверженной длительной неудаче. Альтернативное лечение, которое обычно не применяется, - это перенос сухожилия, который включает перестановку прикрепления мышц к кости. Примеры включают передачу большой грудной мышцы и Процедура Эдена-Ланге.[69]
- Управление лопаточным крылом

Кинезиологический тейп накладывается на лопатки.

Тканевый бандаж для удержания лопаток втянутым, чтобы уменьшить симптомы плеча, такие как боль в ключице.

Скапулопексия между лопаткой и лопаткой, до и после операции. Лопатки фиксируются трансплантатом ахиллова сухожилия, удерживая их во втянутом положении. На правом изображении хорошо видны большие ромбовидные мышцы.



Эпидемиология
В распространенность FSHD колеблется от 1 из 8 333 до 1 из 15 000.[2] Нидерланды сообщают о распространенности 1 из 8 333 после учета недиагностированных.[70] Распространенность в Соединенных Штатах обычно оценивается как 1 из 15 000.[10]
После того, как в 1992 г. стало возможным генетическое тестирование, было установлено, что средняя распространенность составляет около 1 на 20 000, что значительно больше, чем до 1992 г.[71][22][70] Однако 1 из 20 000, вероятно, является заниженной оценкой, поскольку многие с ЛЛПД имеют легкие симптомы и никогда не диагностируются, или они являются братьями и сестрами пострадавших людей и никогда не обращаются за диагнозом.[70]
Не было показано, что раса и этническая принадлежность влияют на частоту или тяжесть ЛЛПД.[10]
Хотя наследование FSHD не показывает склонности к биологическому полу, болезнь проявляется реже у женщин, и даже когда он проявляется у женщин, они в среднем страдают в меньшей степени, чем мужчины.[10] Эстроген подозревается, что это защитный фактор, объясняющий это несоответствие. Одно исследование показало, что эстроген снижает активность DUX4.[72] Однако другое исследование не обнаружило связи между тяжестью заболевания и пожизненным воздействием эстрогена у женщин. То же исследование показало, что прогрессирование болезни не отличалось от периодов гормональных изменений, таких как менархе, беременность и менопауза.[73]
История
Первое описание человека с ЛЛПД в медицинской литературе появляется в отчете о вскрытии трупа. Жан Крювелье в 1852 г.[11][12] В 1868 году Дюшен опубликовал свою основополагающую работу о Мышечная дистрофия Дюшенна, и как часть его дифференциал было описание ЛЛД.[74][12] Сначала в 1874 г., затем в более цитируемой публикации 1884 г. и снова в 1885 г. с изображениями французские врачи Луи Ландузи и Джозеф Дежерин опубликовали подробности болезни, признав ее как отдельную клиническую сущность, и поэтому ЛЛПД иногда называют Болезнь Ландузи-Дежерина.[13][12] В своей статье 1886 года Ландузи и Дежерин обратили внимание на семейную природу расстройства и упомянули, что четыре поколения были затронуты в родстве, которое они исследовали.[75] Формального определения клинических особенностей ЛЛПД не было до 1952 года, когда была изучена большая семья из Юты с ЛЛПД. Примерно с 1980 года возрастающий интерес к ЛЛПД привел к более глубокому пониманию огромной вариабельности заболевания и растущему пониманию генетических и патофизиологических сложностей. К концу 1990-х исследователи наконец начали понимать области хромосомы 4, связанные с ЛЛД.[31]
С момента публикации объединяющей теории в 2010 году исследователи продолжали уточнять свое понимание DUX4. С растущим доверием к этой работе исследователи предложили в 2014 году первую консенсусную точку зрения на патофизиологию заболевания и возможные подходы к терапевтическому вмешательству, основанные на этой модели.[14]
На протяжении многих лет ЛЛПД в разное время называли:
- фациоскапуло-плечевая болезнь[17]
- фасциально-лопаточный[нужна цитата ]
- Болезнь Ландузи-Дежерина[17]
- Синдром Ландузи-Дежерина[75]
- Тип мышечной дистрофии Ландузи-Дежерина[17]
- Синдром Эрба-Ландузи-Дежерина[нужна цитата ]
- Ландузи и Дежерин описывают форму прогрессирующей мышечной атрофии в детстве с характерным поражением лицевых мышц, отличной от псевдогипертрофической (MD Дюшенна) и атрофии спинных мышц у взрослых.[76]
1886
- Ландузи и Дежерин описывают прогрессирующую мышечную атрофию лопаточно-плечевого типа.[77]
1950
- Тайлер и Стивенс изучают 1249 человек из одного единственного рода с ЛЛПД, восходящего к одному предку, и описывают типичный Менделирующее наследование паттерн с полной пенетрантностью и сильно изменчивой экспрессией. Введен термин фациоскапуло-плечевая дистрофия..[78]
1982
- Падберг проводит первые исследования связей, чтобы определить генетический локус за ЛЛПД в его основополагающей диссертации «Лицо-лопаточно-плечевая болезнь».[17]
1987
- Полная последовательность Дистрофин ген (Доктор медицины Дюшенна ) определен.[79]
1991
- Генетический дефект при ЛЛД связан с областью (4q35) возле кончика длинной руки хромосома 4.[80]
1992
- ЛЛПД, как в семейном, так и в de novo дела, оказывается, связано с событием рекомбинации, которое уменьшает размер 4q ЭкоR1 фрагмент до <28 kb (обычно 50–300 kb).[53]
1993
- 4кв. ЭкоБыло обнаружено, что фрагменты R1 содержат тандемное расположение нескольких единиц размером 3,3 т.п.н. (D4Z4), а FSHD связан с наличием <11 единиц D4Z4.[52]
- Исследование семи семей с ЛЛПД обнаруживает доказательства генетическая гетерогенность в ЛЛДП.[81]
1994
- В гетерохроматический структура 4q35 считается фактором, который может влиять на экспрессию FSHD, возможно, через позиционно-эффектное разнообразие.[82]
- Секвенирование ДНК в единицах D4Z4 показывает, что они содержат открытую рамку считывания, соответствующую двум гомеобокс домены, но исследователи пришли к выводу, что D4Z4 вряд ли будет кодировать функциональный транскрипт.[82][83]
1995
- Термины FSHD1A и FSHD1B введены для описания 4q-сцепленных и не-4q-связанных форм заболевания.[84]
1996
1998
- Монозиготные близнецы с совершенно разными клиническими проявлениями FSHD.[15]
1999
- Полное секвенирование единиц D4Z4 4q35 выявляет промоторную область, расположенную на расстоянии 149 п.н. 5 'от открытой рамки считывания для двух гомеобоксов, что указывает на ген, который кодирует белок из 391 аминокислоты (позже исправлен до 424 аминокислотных остатков).[86]), получив имя DUX4.[87]
2001
- Следователи оценили метилирование государственный (гетерохроматин более метилирован, чем эухроматин ) ДНК в 4q35 D4Z4. Экспертиза SmaЯ, MluЯ, МешокII и Eagя рестрикционные фрагменты из нескольких типов клеток, включая скелетные мышцы, не выявило доказательств гипометилирования в клетках пациентов с FSHD1 по сравнению с D4Z4 из непораженных контрольных клеток или по отношению к гомологичным сайтам D4Z4 на хромосома 10. Однако во всех случаях D4Z4 из сперматозоидов был гипометилирован относительно D4Z4 из сперматозоидов. соматический ткани.[88]
2002
- Полиморфный сегмент размером 10 т.п.н. непосредственно дистальнее D4Z4, как обнаружено, существует в двух аллельный формы, обозначенные 4qA и 4qB. FSHD1 связан исключительно с аллелем 4qA.[89]
- Три гена (FRG1, ФРГ2, ANT1 ) расположен в районе центромерный к D4Z4 на хромосоме 4 обнаруживаются в изолированных мышечных клетках людей с FSHD на уровнях, в 10–60 раз превышающих нормальные, что показывает связь между сокращениями D4Z4 и измененной экспрессией генов 4q35.[90]
2003
- Дальнейшее исследование метилирования ДНК в различных рестрикционных фрагментах 4q35 D4Z4 (BsaAя и FseI) показало значительное гипометилирование в обоих сайтах для лиц с FSHD1, носителей генов, не экспрессирующих FSHD, и лиц с фенотипический ЛЛПД относительно здоровых контролей.[91]
2004
- Сокращение области D4Z4 на аллеле 4qB до <38 т.п.н. не вызывает ЛЛД.[92]
2006
- Показано, что у трансгенных мышей со сверхэкспрессией FRG1 развивается тяжелая миопатия.[93]
2007
- Обнаружено, что открытая рамка считывания DUX4 сохраняется в геноме приматов более 100 миллионов лет, что подтверждает вероятность того, что она кодирует необходимый белок.[94]
- Исследователи идентифицируют мРНК DUX4 при первичной ЛЛД. миобласты и идентифицировать в клетках, трансфицированных D4Z4, белок DUX4, сверхэкспрессия которого вызывает гибель клеток.[86]
- DUX4 мРНК и экспрессия белка, как сообщается, увеличивается в миобластах от пациентов с ЛЛПД по сравнению с неповрежденной контрольной группой. Стабильная мРНК DUX4 транскрибируется только с наиболее дистальной единицы D4Z4, которая использует интрон и полиаденилирование сигнал обеспечивается фланкирующей областью pLAM. Белок DUX4 идентифицирован как фактор транскрипции, и данные свидетельствуют о том, что сверхэкспрессия DUX4 связана с увеличением целевого парно-подобная транскрипция гомеодомена фактор 1 (PITX1 ).[95]
2009
- Термины FSHD1 и FSHD2 введены для описания генетических форм, связанных с делецией D4Z4 и не связанных с делецией D4Z4, соответственно. В FSHD1 гипометилирование ограничивается коротким аллелем 4q, тогда как FSHD2 характеризуется гипометилированием как 4q, так и обоих аллелей 10q.[96]
- Сплайсинг и расщепление терминала (большинство теломерный ) Транскрипт 4q D4Z4 DUX4 в первичных миобластах и фибробластах от пациентов с ЛЛД приводит к генерации множественных РНК, в том числе малых некодирующие РНК, антисмысловые РНК и кэпированные мРНК как новые кандидаты в патофизиология ЛЛДП.[97]
2010
- Установлена объединяющая генетическая модель ЛПД: сокращения D4Z4 вызывают ЛПД только в контексте аллеля 4qA из-за стабилизации DUX4 Транскрипт РНК, позволяющий DUX4 выражение.[6] Несколько организаций, включая Нью-Йорк Таймс подчеркнул это исследование[98] (Видеть Общество FSHD ).
Доктор Фрэнсис Коллинз, который курировал первую последовательность Человеческий геном с Национальные институты здоровья заявил:[98]
"Если бы мы думали о сборнике лучших хитов генома, это было бы в списке",
Даниэль Перес, соучредитель Общества FSHD, приветствовал новые результаты, сказав:[нужна цитата ]
«Это долгожданное объяснение точного биологического действия [ЛЛПД]»
MDA заявило, что:[нужна цитата ]
"Теперь идет охота за белками или генетическими инструкциями (РНК ) вызывают проблемы с мышечной тканью при ЛЛПД ".
Один из соавторов отчета, Сильвер ван дер Маарель из Лейденского университета, заявил, что[нужна цитата ]
«Удивительно осознавать, что долгое и разочаровывающее путешествие, длившееся почти два десятилетия, теперь завершилось идентификацией одного небольшого варианта ДНК, который различается между пациентами и людьми, не страдающими этим заболеванием. Наконец-то у нас есть цель, к которой мы можем идти ».
- DUX4 активно транскрибируется в биоптатах скелетных мышц и первичных миобластах. Клетки, пораженные FSHD, продуцируют полноразмерный транскрипт, DUX4-fl, тогда как альтернативный сплайсинг у здоровых людей приводит к образованию более короткого, усеченного 3'-транскрипта (DUX4-s). Низкая общая экспрессия обоих транскриптов в мышцах объясняется относительно высокой экспрессией в небольшом количестве ядер (~ 1 из 1000). Более высокие уровни экспрессии DUX4 в семенниках человека (примерно в 100 раз выше, чем в скелетных мышцах) предполагают роль DUX4 в развитии человека. Показано, что более высокие уровни DUX4-s (по сравнению с DUX4-fl) коррелируют с большей степенью метилирования DUX-4 H3K9me3.[5]
2012
- Некоторые экземпляры FSHD2 связаны с мутациями в гене SMCHD1 на хромосома 18, и установлено генетическое / механистическое пересечение FSHD1 и FSHD2.[40]
- Распространенность FSHD-подобных делеций D4Z4 на пермиссивных аллелях значительно выше, чем распространенность FSHD в общей популяции, что ставит под сомнение критерии молекулярной диагностики FSHD.[99]
- При экспрессии в первичных миобластах DUX4-fl действует как активатор транскрипции, вызывая более чем 3-кратное изменение экспрессии 710 генов.[100] Последующее исследование с использованием большего количества образцов выявило экспрессию DUX4-fl в миогенных клетках и мышечной ткани здоровых родственников пациентов с ЛЛД. per se, недостаточно, чтобы вызвать патологию, и что дополнительные модификаторы являются детерминантами прогрессирования заболевания.[101]
- Предложен механизм DBE-T (транскрипт регуляторного элемента D4Z4), приводящий к дерепрессии генов 4q35.[36]
2013
- Показано, что мутации в SMCHD1 увеличивают тяжесть FSHD1.[44]
- Трансгенные мыши несущие массивы D4Z4 от аллеля FSHD1 (с 2,5 единицами D4Z4), хотя и не имеют очевидного FSHD-подобного фенотипа скелетных мышц, как обнаружено, повторяют важные паттерны генетической экспрессии и эпигенетический особенности ЛЛД.[102]
2014
- DUX4-fl и нижестоящие гены-мишени экспрессируются в биоптатах скелетных мышц и полученных из биопсии клетках плодов с FSHD-подобными матрицами D4Z4, что указывает на то, что молекулярные маркеры FSHD уже экспрессируются во время развития плода.[103]
- Исследователи «рассматривают, как вклад многих лабораторий в течение многих лет привел к пониманию принципиально нового механизма болезней человека» и формулируют, как объединяющая генетическая модель и последующие исследования представляют собой «поворотную точку в исследованиях ЛЛПД, переходя область от открытий. -ориентированные исследования на трансляционные исследования, направленные на разработку методов лечения, основанных на надежной модели патофизиологии болезни ». Они описывают консенсусный механизм патофизиологии ЛЛД как «неэффективное опосредованное повторами эпигенетическое подавление массива макросателлитных повторов D4Z4 на хромосоме 4, приводящее к неоднородной экспрессии DUX4 ретроген, кодирующий фактор транскрипции двойного гомеобокса, в скелетных мышцах ». [14]
Прошлые фармацевтические разработки
Ранние испытания лекарств, до патогенеза, включающего DUX4 были обнаружены, не были нацелены и по большей части безуспешны.[104] Большинство соединений были испытаны на основе увеличения мышечной массы или уменьшения воспаления.[104] К лекарствам, которые не показали эффективности, относятся:
- Преднизон, стероид, был испытан из-за его терапевтического эффекта при мышечной дистрофии Дюшенна.[105]
- Устный альбутерол, а β2 агонист Хотя в клинических испытаниях он улучшил мышечную массу и некоторые показатели силы, он не улучшил общую силу или функцию.[106][107][108] Интересно, что после того, как DUX4 был идентифицирован как неотъемлемая часть патофизиологии FSHD, скрининг лекарственных средств показал, что агонисты β2 снижают экспрессию DUX4.[109]
- Дилтиазем Блокатор кальциевых каналов был испытан при ЛЛПД на основании отдельных сообщений о его полезности и теории о том, что нарушение регуляции кальция может играть роль в гибели мышечных клеток (это было до идентификации DUX4 как части патофизиологии).[110]
- MYO-029 (Стамулумаб ) был разработан для стимулирования роста мышц. Это антитело, которое подавляет миостатин, белок, подавляющий рост мышечной ткани.[111]
- ACE-083 - ингибитор TGF-β, разработанный для стимулирования роста мышц.[112]
Общество и культура
- в Amazon Видео серии Человек в высоком замке, Обергруппенфюрер У сына Джона Смита, Томаса, диагностирован синдром Ландузи-Дежерина.
- В биографии Стюарт: Жизнь в обратном направлении, главный герой пострадал от ЛЛСД.
- Крис Каррино, радио голос Бруклин Нетс, подвержен FSHD. Он создал Фонд Криса Каррино для ЛЛБД.
Общество FSHD
В 1991 г. Общество FSHD (до 2019 года называлось «Общество FSH»)[113] была основана двумя людьми с FSHD, Дэниелом Пересом и Стивеном Якобсеном. Общество FSHD собрало финансирование для предоставления посевных грантов для исследований FSHD, выступило за стандартизацию названия болезни как фасциально-лопаточно-плечевая мышечная дистрофия и FSHDи соавтором Закон о MD-CARE, принятый в 2001 году закон, который впервые санкционировал федеральные ресурсы, в том числе Национальные институты здоровья финансирование при всех мышечных дистрофиях. Общество FSHD превратилось в крупнейшую в мире массовую организацию, выступающую за обучение пациентов, а также научные и медицинские исследования.[114]
FSHD-ЕВРОПА
В 2009 году европейские ассоциации основали FSHD-EUROPE.[115]
Направления исследований
На основе модели консенсуса патофизиологии исследователи предлагают четыре подхода к терапевтическому вмешательству:[14]
- усилить эпигенетическую репрессию D4Z4
- нацеливать мРНК DUX4, включая изменение сплайсинга или полиаденилирования;
- блокировать активность белка DUX4
- подавляют индуцированный DUX4 процесс или процессы, которые приводят к патологии.
Текущие фармацевтические разработки
- Лосмапимод, селективный ингибитор p38α / β митоген-активируемые протеинкиназы, был идентифицирован Fulcrum Therapeutics как мощный супрессор DUX4 in vitro.[116] Клинические испытания фазы IIb начались в июле 2019 года и, как ожидается, завершатся в августе 2020 года.[117]
- Антисмысловые нуклеотиды, направленные против матричной РНК DUX4, находятся на доклинической стадии. Было показано, что антисмысловые нуклеотиды уменьшают DUX4 и подавляют гены-мишени DUX4 с небольшими побочными эффектами. В настоящее время проблема заключается в доставке нуклеотидов в мышечные клетки; эти антисмысловые нуклеотиды обладают плохой способностью проникать в мышцу.[2]
- Генная терапия состоящие из микроРНК (миРНК), направленных против DUX4, доставляемых вирусными векторами, находятся на доклинической стадии. В моделях FSHD на мышах было показано, что miRNAs уменьшают DUX4, защищают от мышечной патологии и предотвращают потерю силы захвата.[2]
Возможное фармацевтическое развитие
- Подавление гиалуроновая кислота (HA) путь является потенциальной терапией. Одно исследование показало, что многие DUX4-индуцированные молекулярные патологии опосредуются передачей сигналов HA, и ингибирование биосинтеза HA с помощью 4-метилумбеллиферона предотвращает эти молекулярные патологии.[118]
- Было показано, что ингибирование P300 подавляет вредные эффекты DUX4[119]
- Ингибиторы БЭТ было показано, что снижает экспрессию DUX4.[109]
- Казеин киназа 1 (CK1) ингибиторы были идентифицированы голландской фармацевтической компанией Facio Therapies как репрессоры DUX4 выражение. Facio Therapies утверждает, что ингибирование CK1 не нарушает слияние мышечных трубок, в отличие от ингибиторов BET, p38 MAPK ингибиторы и β2 агонисты.[120][121]
- Антиоксиданты потенциально могут уменьшить эффекты ЛЛПД. Одно исследование показало, что Витамин С, витамин Е, глюконат цинка, и селенометионин добавка увеличивала выносливость и силу четырехглавой мышцы, но не оказывала существенного влияния на ходьбу.[122] Необходимы дальнейшие исследования.[2]
Критерии оценки
Способы измерения заболевания важны для оценки эффективности лекарств в клинических испытаниях.
- Электрическая импедансная миография изучается как способ измерения повреждения мышц.[2]
- Качество жизни можно измерить с помощью анкет, таких как Индекс здоровья FSHD.[123][2]
- МРТ мышц полезна для оценки всех мышц тела. Мышцы могут быть оценены в зависимости от степени инфильтрации жира.[2]
Рекомендации
- ^ Рикард, Аманда; Петек, Лиза; Миллер, Дэниел (5 августа 2015 г.). «Эндогенная экспрессия DUX4 в мышечных трубках FSHD достаточна, чтобы вызвать гибель клеток и нарушить пути сплайсинга РНК и миграции клеток». Гм. Мол. Genet. 24 (20): 5901–14. Дои:10.1093 / hmg / ddv315. ЧВК 4581613. PMID 26246499. Получено 10 сентября, 2015.
- ^ а б c d е ж грамм час я j k л м п о п q р s т ты v ш Икс у z аа ab ac объявление ае аф аг ах ай эй ак аль являюсь Вагнер, Кэтрин Р. (декабрь 2019 г.). «Лице-лопаточно-плечевые мышечные дистрофии». КОНТИНУУМ: обучение в неврологии на протяжении всей жизни. 25 (6): 1662–1681. Дои:10.1212 / CON.0000000000000801. PMID 31794465. S2CID 208531681.
- ^ а б Tawil, R; Ван дер Маарель, С.М. (июль 2006 г.). «Лице-лопаточно-плечевая мышечная дистрофия» (PDF). Мышцы и нервы. 34 (1): 1–15. Дои:10.1002 / mus.20522. PMID 16508966. S2CID 43304086.
- ^ а б Кумар, Винай; Аббас, Абул; Астер, Джон, ред. (2018). Базовая патология Роббинса (Десятое изд.). Филадельфия, Пенсильвания: Эльзевир. п. 844. ISBN 978-0-323-35317-5.
- ^ а б c Снайдер, L; Geng, LN; Lemmers, RJ; Киба, М; Посуда, CB; Нельсон, AM; Tawil, R; Филиппова, Г.Н.; van der Maarel, SM; Tapscott, SJ; Миллер, Д.Г. (28 октября 2010 г.). «Фациоскапуло-плечевая дистрофия: неполное подавление ретротранспозированного гена». PLOS Genetics. 6 (10): e1001181. Дои:10.1371 / journal.pgen.1001181. ЧВК 2965761. PMID 21060811.
- ^ а б c d е ж грамм час я j k л м Lemmers RJ, van der Vliet PJ, Klooster R, Sacconi S, Camaño P, Dauwerse JG, Snider L, Straasheijm KR, van Ommen GJ, Padberg GW, Miller DG, Tapscott SJ, Tawil R, Frants RR, van der Maarel SM ( 19 августа 2010 г.). "Объединяющая генетическая модель лицево-капуло-плечевой мышечной дистрофии" (PDF). Наука. 329 (5999): 1650–3. Bibcode:2010Sci ... 329.1650L. Дои:10.1126 / science.1189044. HDL:1887/117104. ЧВК 4677822. PMID 20724583. Архивировано из оригинал (PDF) на 2014-06-05.
- ^ Lemmers RJ, Wohlgemuth M, van der Gaag KJ, et al. (Ноябрь 2007 г.). «Конкретные вариации последовательности в области 4q35 связаны с фасциально-лопаточно-плечевой мышечной дистрофией». Являюсь. J. Hum. Genet. 81 (5): 884–94. Дои:10.1086/521986. ЧВК 2265642. PMID 17924332.
- ^ а б c d е ж грамм час Мул, Карлиен; Ласше, Саския; Voermans, Nicol C; Падберг, Джордж В; Хорлингс, Корин Г.К .; ван Энгелен, Baziel GM (июнь 2016 г.). «Что в названии? Клинические особенности фасциально-плечевой мышечной дистрофии». Практическая неврология. 16 (3): 201–207. Дои:10.1136 / Practneurol-2015-001353. PMID 26862222. S2CID 4481678.
- ^ а б Эрен, Илкер; Бирсель, Ольгар; Озтоп Чакмак, Озгюр; Аслангер, Айса; Гюрсой Оздемир, Ясемин; Ераслан, Серполь; Кайсерили, Хюля; Oflazer, Piraye; Демирхан, Мехмет (май 2020 г.). «Новая система определения стадии инвалидности плеча для лопаточно-грудного артродеза у пациентов с фациально-лопаточной дистрофией». Ортопедия и травматология: хирургия и исследования. 106 (4): 701–707. Дои:10.1016 / j.otsr.2020.03.002. PMID 32430271.
- ^ а б c d Statland, JM; Тавил, Р. (декабрь 2016 г.). «Лице-лопаточно-плечевая мышечная дистрофия». Continuum (Миннеаполис, Миннесота). 22 (6, Заболевания мышц и нервно-мышечного соединения): 1916–1931. Дои:10.1212 / CON.0000000000000399. ЧВК 5898965. PMID 27922500.
- ^ а б Крювелье, Дж. (1852–1853). "Mémoire sur la paralysie musculaire atrophique". Bulletins de l'Académie de Médecine. 18: 490–502, 546–583.
- ^ а б c d е Роджерс, Марк Т. (2004). «Фациоскапуло-плечевая мышечная дистрофия: исторический фон и обзор литературы». В Упадхьяе, Мине; Купер, Дэвид Н. (ред.). Фасциально-плечевая мышечная дистрофия ЛЛД: клиническая медицина и молекулярная клеточная биология. Издательство BIOS Scientific. ISBN 1-85996-244-0.
- ^ а б Ландузи, Л .; Дежерин, Дж. (1885). Ландузи, Л .; Лепин, Р. (ред.). «Прогрессирующая атрофическая миопатия (миопатия без нейропатии, дебютант необычной для детей)». Revue de Médecine (На французском). Феликс Алкан. 5: 253–366. Получено 19 мая 2020.
- ^ а б c d е ж грамм час Тавил, Раби; van der Maarel, SM; Тапскотт, SJ (10 июня 2014 г.). «Фациоскапуло-плечевая дистрофия: путь к консенсусу по патофизиологии». Скелетные мышцы. 4 (1): 12. Дои:10.1186/2044-5040-4-12. ЧВК 4060068. PMID 24940479.
- ^ а б Tupler, R; Барбиерато, L; и другие. (Сентябрь 1998 г.). «Идентичная мутация de novo в локусе D4F104S1 у монозиготных близнецов мужского пола, пораженных фациоскапуло-плечевой мышечной дистрофией (FSHD) с различным клиническим проявлением». Журнал медицинской генетики. 35 (9): 778–783. Дои:10.1136 / jmg.35.9.778. ЧВК 1051435. PMID 9733041.
- ^ Tawil, R; Сторвик, Д.; Фисби, TE; Weiffenbach, B; Григгс, Р.К. (февраль 1993 г.). «Чрезвычайная вариабельность экспрессии у монозиготных близнецов с мышечной дистрофией ФСГ». Неврология. 43 (2): 345–8. Дои:10.1212 / wnl.43.2.345. PMID 8094896. S2CID 44422140.
- ^ а б c d е Падберг, GW (1982-10-13). Лице-лопаточно-плечевое заболевание (Тезис). Лейденский университет.
- ^ а б Райкен, штат Нью-Хэмпшир; van der Kooi, EL; Хендрикс, JC; ван Асселдонк, Р.Дж.; Падберг, GW; Geurts, AC; ван Энгелен, Б.Г. (декабрь 2014 г.). «Визуализация скелетных мышц при фасциально-плечевой мышечной дистрофии, модели и асимметрии поражения отдельных мышц». Нервно-мышечные расстройства. 24 (12): 1087–96. Дои:10.1016 / j.nmd.2014.05.012. PMID 25176503. S2CID 101093.
- ^ Пандья, Шри; Эйхингер, Кейт. «Физиотерапия при лицево-плечевой мышечной дистрофии» (PDF). Общество ЛЛСД. Архивировано из оригинал (PDF) 14 апреля 2020 г.. Получено 14 апреля 2020.
- ^ а б c Упадхьяя, Мина; Купер, Дэвид, ред. (Март 2004 г.). FSHD Facioscapulohumeral Muscular Dystrophy: Клиническая медицина и молекулярно-клеточная биология. Издательство BIOS Scientific. ISBN 0203483677.
- ^ Тревизан, CP; Пасторелло, E; Tomelleri, G; Верчелли, L; Бруно, К; Скаполан, S; Сицилиано, G; Комаккио, Ф (декабрь 2008 г.). «Фациоскапуло-плечевая мышечная дистрофия: потеря слуха и другие атипичные особенности пациентов с большими делециями 4q35». Европейский журнал неврологии. 15 (12): 1353–8. Дои:10.1111 / j.1468-1331.2008.02314.x. PMID 19049553. S2CID 26276887.
- ^ а б c Падберг, Джордж У. (2004). «Лице-лопаточно-плечевая мышечная дистрофия: опыт клинициста». В Упадхьяе, Мина; Купер, Дэвид Н. (ред.). FSHD Facioscapulohumeral Muscular Dystrophy: Клиническая медицина и молекулярно-клеточная биология. Издательство BIOS Scientific. ISBN 1-85996-244-0.
- ^ а б c Таска, G; Монфорте, М; Iannaccone, E; Laschena, F; Оттавиани, П; Leoncini, E; Boccia, S; Галлуцци, G; Pelliccioni, M; Masciullo, M; Frusciante, R; Меркурий, Е; Риччи, Э (2014). «Визуализация верхнего пояса при фациоскапуло-плечевой мышечной дистрофии». PLOS ONE. 9 (6): e100292. Bibcode:2014PLoSO ... 9j0292T. Дои:10.1371 / journal.pone.0100292. ЧВК 4059711. PMID 24932477.
- ^ а б c Геревини, С; Скарлато, М; Maggi, L; Cava, M; Калиендо, G; Пасаниси, Б; Фалини, А; Previtali, SC; Моранди, Л. (март 2016 г.). «Результаты МРТ мышц при фасциально-плечевой мышечной дистрофии». Европейская радиология. 26 (3): 693–705. Дои:10.1007 / s00330-015-3890-1. PMID 26115655. S2CID 24650482.
- ^ Пандья, Шри; Кинг, Венди М; Тавил, Раби (1 января 2008 г.). «Лице-лопаточно-плечевая дистрофия». Физиотерапия. 88 (1): 105–113. Дои:10.2522 / ptj.20070104. PMID 17986494.
- ^ а б c Mair, D; Huegens-Penzel, M; Кресс, Вт; Рот, С; Ферберт, А (2017). «Вовлечение мышц ног в лицево-плечевую мышечную дистрофию: сравнение 1-го и 2-го типов фасциально-плечевой мышечной дистрофии». Европейская неврология. 77 (1–2): 32–39. Дои:10.1159/000452763. PMID 27855411. S2CID 25005883.
- ^ а б c Olsen, DB; Гидеон, П; Jeppesen, TD; Виссинг, Дж. (Ноябрь 2006 г.).«Поражение мышц ног при фациоскапуло-плечевой мышечной дистрофии, оцененное с помощью МРТ». Журнал неврологии. 253 (11): 1437–41. Дои:10.1007 / s00415-006-0230-z. PMID 16773269. S2CID 19421344.
- ^ Линднер, Мориц; Хольц, Фрэнк Дж. Чарбель Исса, Питер (2016-04-27). «Спонтанное разрешение сосудистых аномалий сетчатки и отека макулы при фациоскапуло-плечевой мышечной дистрофии». Клиническая и экспериментальная офтальмология. 44 (7): 627–628. Дои:10.1111 / ceo.12735. ISSN 1442-6404. PMID 26933772. S2CID 204996841.
- ^ Вольгемут М., ван дер Кой Э.Л., ван Кестерен Р.Г., ван дер Маарель С.М., Падберг Г.В. (2004). «Вентиляционная поддержка при фасциально-плечевой мышечной дистрофии». Неврология. 63 (1): 176–8. CiteSeerX 10.1.1.543.2968. Дои:10.1212 / 01.wnl.0000133126.86377.e8. PMID 15249635. S2CID 31335126.
- ^ а б c d е ж грамм Lemmers, Richard J.L.F .; О'Ши, Сюзанна; Падберг, Джордж В .; Лант, Питер У .; ван дер Маарель, Сильвер М. (май 2012 г.). «Рекомендации по передовой практике генетической диагностики лицево-плечевой мышечной дистрофии: семинар, 9 июня 2010 г., LUMC, Лейден, Нидерланды». Нервно-мышечные расстройства. 22 (5): 463–470. Дои:10.1016 / j.nmd.2011.09.004. PMID 22177830. S2CID 39898514.
- ^ а б c d Невозможные вещи: через зеркало с исследователями дистрофии ФСГ, Маргарет Валь, MDA, Журнал Quest, Том 14, № 2, март – апрель 2007 г.
- ^ Диксит М., Анссо Э., Тассин А. и др. (Ноябрь 2007 г.). «DUX4, ген-кандидат фациоскапуло-плечевой мышечной дистрофии, кодирует активатор транскрипции PITX1». Proc. Natl. Акад. Sci. СОЕДИНЕННЫЕ ШТАТЫ АМЕРИКИ. 104 (46): 18157–62. Bibcode:2007PNAS..10418157D. Дои:10.1073 / pnas.0708659104. ЧВК 2084313. PMID 17984056.
- ^ White, J.A .; McAlpine, P.J .; Antonarakis, S .; Cann, H .; Eppig, J.T .; Фрейзер, К .; Frezal, J .; Lancet, D .; Nahmias, J .; Pearson, P .; Peters, J .; Scott, A .; Scott, H .; Spurr, N .; Talbot, C .; Пови, С. (октябрь 1997 г.). "НОМЕНКЛАТУРА". Геномика. 45 (2): 468–471. Дои:10.1006 / geno.1997.4979. PMID 9344684.
- ^ Fasman, KH; Летовский, С.И. Коттингем, RW; Кингсбери, ДТ (1 января 1996 г.). «Усовершенствования базы данных генома человека GDB». Исследования нуклеиновых кислот. 24 (1): 57–63. Дои:10.1093 / nar / 24.1.57. ЧВК 145602. PMID 8594601.
- ^ Himeda, CL; Джонс, Польша (31 августа 2019 г.). "Генетика и эпигенетика фасциально-плечевой мышечной дистрофии". Ежегодный обзор геномики и генетики человека. 20: 265–291. Дои:10.1146 / annurev-genom-083118-014933. PMID 31018108.
- ^ а б Cabianca, DS; Casa, Casa; Bodega, B; и другие. (11 мая 2012 г.). «Длинная нкРНК связывает изменение числа копий с эпигенетическим переключателем поликомб / триторакс при мышечной дистрофии ЛЛПД». Клетка. 149 (4): 819–831. Дои:10.1016 / j.cell.2012.03.035. ЧВК 3350859. PMID 22541069.
- ^ а б c d е ж грамм Sacconi, S; Бриан-Суло, А; Gros, M; Baudoin, C; Леммерс, RJLF; Рондо, S; Lagha, N; Nigumann, P; Cambieri, C; Пума, А; Чапон, F; Стойкович, Т; Флакон, C; Bouhour, F; Цао, М; Pegoraro, E; Petiot, P; Бехин, А; Marc, B; Эймар, Б; Эчаниз-Лагуна, А; Laforet, P; Salviati, L; Jeanpierre, M; Кристофари, G; ван дер Маарель, С.М. (7 мая 2019 г.). «FSHD1 и FSHD2 образуют континуум болезни». Неврология. 92 (19): e2273 – e2285. Дои:10.1212 / WNL.0000000000007456. ЧВК 6537132. PMID 30979860.
- ^ Tupler, R; Берардинелли, А; Барбиерато, L; Франц, Р; Hewitt, JE; Lanzi, G; Maraschio, P; Тьеполо, Л. (май 1996 г.). «Моносомия дистального 4q не вызывает фациоскапуло-плечевой мышечной дистрофии». Журнал медицинской генетики. 33 (5): 366–70. Дои:10.1136 / jmg.33.5.366. ЧВК 1050603. PMID 8733044.
- ^ Росси М., Риччи Э., Колантони Л. и др. (2007). «Область фасциоскапуло-плечевой мышечной дистрофии на 4-м квартале и гомологичный локус на 10-м квартале развивались независимо под различным эволюционным давлением». BMC Med. Genet. 8: 8. Дои:10.1186/1471-2350-8-8. ЧВК 1821008. PMID 17335567.
- ^ а б c Lemmers, RJ; Tawil, R; Петек, Л.М.; и другие. (Декабрь 2012 г.). «Дигенное наследование мутации SMCHD1 и FSHD-пермиссивного аллеля D4Z4 вызывает фациоскапуло-плечевую мышечную дистрофию 2 типа». Природа Генетика. 44 (12): 1370–1374. Дои:10.1038 / ng.2454. ЧВК 3671095. PMID 23143600.
- ^ ван ден Богаард, ML; Леммерс, RJLF; Балог, Дж; Вольгемут, М; Ауранен, М; Mitsuhashi, S; van der Vliet, PJ; Страашейм, КР; ван ден Аккер, RFP; Kriek, M; Лоренс-Бик, MEY; Раз, В; van Ostaijen-Ten Dam, MM; Ханссон, КБМ; van der Kooi, EL; Киуру-Энари, S; Удд, В; ван Тол, MJD; Нишино, I; Tawil, R; Tapscott, SJ; ван Энгелен, BGM; ван дер Маарель, С.М. (5 мая 2016 г.). «Мутации в DNMT3B модифицируют эпигенетическую репрессию повтора D4Z4 и проникновение лицево-лопаточно-плечевой дистрофии». Американский журнал генетики человека. 98 (5): 1020–1029. Дои:10.1016 / j.ajhg.2016.03.013. ЧВК 4863565. PMID 27153398.
- ^ а б Джонсон, штат Невада; Статленд, JM (7 мая 2019 г.). «FSHD1 или FSHD2: вот в чем вопрос: ответ: это все просто FSHD». Неврология. 92 (19): 881–882. Дои:10.1212 / WNL.0000000000007446. PMID 30979855. S2CID 111390628.
- ^ а б c Хаманака, Кохей; Шикрова, Дарина; Мицухаси, Сатоми; Масуда, Хироки; Сэкигучи, Юкари; Сугияма, Атсухико; Сибуя, Кадзумото; Lemmers, Richard J.L.F .; Гуссенс, Ремко; Огава, Мегуму; Нагао, Кодзи; Обусе, Чикаши; Ногучи, Сатору; Хаяси, Юкико К .; Кувабара, Сатоши; Балог, Юдит; Нишино, Ичизо; ван дер Маарель, Сильвер М. (28 мая 2020 г.). «Гомозиготный бессмысленный вариант, ассоциированный с фасциально-плечевой мышечной дистрофией». Неврология. 94 (23): e2441 – e2447. Дои:10.1212 / WNL.0000000000009617. ЧВК 7455367. PMID 32467133. S2CID 218982743.
- ^ а б Sacconi, S; Lemmers, RJ; Балог, Дж; и другие. (3 октября 2013 г.). «Ген FSHD2 SMCHD1 является модификатором тяжести заболевания в семьях, затронутых FSHD1». Американский журнал генетики человека. 93 (4): 744–751. Дои:10.1016 / j.ajhg.2013.08.004. ЧВК 3791262. PMID 24075187.
- ^ а б Lim, KRQ; Нгуен, Q; Йокота, Т (22 января 2020 г.). «Передача сигналов DUX4 в патогенезе фациоскапуло-плечевой мышечной дистрофии». Международный журнал молекулярных наук. 21 (3): 729. Дои:10.3390 / ijms21030729. ЧВК 7037115. PMID 31979100.
- ^ а б Боснаковский, Дарко; Shams, Ahmed S .; Юань, Се; да Силва, Meiricris T .; Ener, Элизабет Т .; Бауманн, Кори У .; Линдси, Ангус Дж .; Верма, Маянк; Асакура, Ацуши; Lowe, Dawn A .; Киба, Майкл (6 апреля 2020 г.). «Транскрипционные и цитопатологические признаки ЛПД у мышей с хронической экспрессией DUX4». Журнал клинических исследований. 130 (5): 2465–2477. Дои:10.1172 / JCI133303. ЧВК 7190912. PMID 32250341.
- ^ Лек, Анджела; Чжан, Юаньфань; Woodman, Keryn G .; Хуанг, Шушу; DeSimone, Alec M .; Коэн, Джастин; Хо, Винсент; Коннер, Джеймс; Мид, Лилиан; Кодани, Эндрю; Пакула, Анна; Санджана, Невилл; Кинг, Оливер Д .; Джонс, Питер Л .; Вагнер, Кэтрин Р .; Лек, Монкол; Кункель, Луи М. (25 марта 2020 г.). «Применение полногеномных экранов CRISPR-Cas9 для терапевтических открытий при фациоскапуло-плечевой мышечной дистрофии». Научная трансляционная медицина. 12 (536): eaay0271. Дои:10.1126 / scitranslmed.aay0271. ЧВК 7304480. PMID 32213627.
- ^ Страфелла, Клаудиа; Капуто, Валерио; Галота, Розария Мария; Камполи, Джулия; Бакс, Кристина; Колантони, Лука; Миноцци, Джульетта; Орсини, Кьяра; Политано, Луиза; Таска, Джорджио; Новелли, Джузеппе; Риччи, Энцо; Джардина, Эмилиано; Каселла, Рафаэлла (1 декабря 2019 г.). «Вариабельность гена SMCHD1 у пациентов с ЛЛПД: свидетельство новых мутаций». Молекулярная генетика человека. 28 (23): 3912–3920. Дои:10.1093 / hmg / ddz239. ЧВК 6969370. PMID 31600781.
- ^ Зампатти, S; Colantoni, L; Strafella, C; Галота, РМ; Капуто, V; Камполи, G; Pagliaroli, G; Карбони, S; Мела, Дж; Пекони, С; Gambardella, S; Cascella, R; Джардина, Э (май 2019 г.). «Молекулярная диагностика лицево-плечевой мышечной дистрофии (ЛЛД): от традиционных технологий к эпохе NGS». Нейрогенетика. 20 (2): 57–64. Дои:10.1007 / s10048-019-00575-4. PMID 30911870. S2CID 85495566.
- ^ Киношита, июнь (11 марта 2020 г.). «Генетическое тестирование на ЛЛПД - новый рубеж». Общество FSHD. Архивировано из оригинал 8 апреля 2020 г.. Получено 8 апреля 2020.
- ^ Васале, Дж; Боярин, Ф; Джоксон, М; Сулькова, В; Чан, П; Лиакват, К; Хоффман, C; Месервей, М; Чанг, я; Цао, Д; Хенсли, К; Лю, Y; Оуэн, Р. Браастад, К; Солнце, Вт; Walrafen, P; Komatsu, J; Wang, JC; Бенсимон, А; Ангиано, А; Яремко, М; Ван, З; Батиш, S; Стром, С; Хиггинс, Дж (декабрь 2015 г.). «Молекулярное расчесывание по сравнению с саузерн-блоттингом для измерения сокращений D4Z4 при ЛЛД». Нервно-мышечные расстройства. 25 (12): 945–51. Дои:10.1016 / j.nmd.2015.08.008. PMID 26420234. S2CID 6871094.
- ^ а б van Deutekom, JC; Wijmenga, C; van Tienhoven, EA; и другие. (Декабрь 1993 г.). «Реаранжировки ДНК, связанные с FSHD, происходят из-за делеций целых копий тандемно повторяющейся единицы размером 3,2 т.п.н.». Молекулярная генетика человека. 2 (12): 2037–2042. Дои:10.1093 / hmg / 2.12.2037. PMID 8111371.
- ^ а б Wijmenga, C; Hewitt, JE; Sandkuijl, LA; и другие. (Сентябрь 1992 г.). «Перестройки ДНК в хромосоме 4q, связанные с фациоскапуло-плечевой мышечной дистрофией». Природа Генетика. 2 (1): 26–30. Дои:10.1038 / ng0992-26. PMID 1363881. S2CID 21940164.
- ^ Франц, Rune R .; Sandkuijl, Lodewijk A .; van der Maarel, Silvere M .; Падберг, Джордж У. (2004). «Картирование гена FSHD и открытие патогномоничной делеции». В Упадхьяе, Мина; Купер, Дэвид Н. (ред.). FSHD Facioscapulohumeral Muscular Dystrophy: Клиническая медицина и молекулярно-клеточная биология. Издательство BIOS Scientific. ISBN 1-85996-244-0.
- ^ Информационный бюллетень FSHD В архиве 2006-03-06 на Wayback Machine, MDA, 11/1/2001
- ^ а б Voet, N; Bleijenberg, G; Хендрикс, Дж; де Гроот, я; Падберг, G; ван Энгелен, B; Геуртс, А (18 ноября 2014 г.). «Как аэробные упражнения, так и когнитивно-поведенческая терапия снижают хроническую усталость при ЛЛПД: рандомизированное контролируемое исследование». Неврология. 83 (21): 1914–22. Дои:10.1212 / WNL.0000000000001008. PMID 25339206. S2CID 25382403.
- ^ а б Янссен, Б; Voet, N; Geurts, A; ван Энгелен, B; Heerschap, A (3 мая 2016 г.). «Количественная МРТ выявляет замедленную жировую инфильтрацию в мышцах активных пациентов с ЛЛПД». Неврология. 86 (18): 1700–7. Дои:10.1212 / WNL.0000000000002640. PMID 27037227. S2CID 11617226.
- ^ Voet, Nicoline Bm; van der Kooi, Elly L .; ван Энгелен, Базиэль Гм; Геуртс, Александр Ч (6 декабря 2019). «Силовые тренировки и аэробные упражнения при мышечных заболеваниях». Кокрановская база данных систематических обзоров. 12: CD003907. Дои:10.1002 / 14651858.CD003907.pub5. ISSN 1469-493X. ЧВК 6953420. PMID 31808555.
- ^ а б Tawil, R; Mah, JK; Бейкер, S; Вагнер, КР; Райан, ММ; Сиднейский семинар, участники. (Июль 2016 г.). "Рекомендации по клинической практике при фасциально-плечевой мышечной дистрофии Сидней, Австралия, 21 сентября 2015 г.". Нервно-мышечные расстройства. 26 (7): 462–71. Дои:10.1016 / j.nmd.2016.03.007. PMID 27185458.
- ^ «Информация для пациентов и их семей - Центр Ричарда Филдса по дистрофии ФСГ (FSHD) и нейромышечных исследований - Медицинский центр Университета Рочестера». www.urmc.rochester.edu. Архивировано из оригинал 14 ноября 2019 г.. Получено 14 апреля 2020.
- ^ Априле, I; Bordieri, C; Gilardi, A; Lainieri Milazzo, M; Руссо, G; Де Сантис, Ф; Frusciante, R; Iannaccone, E; Erra, C; Ricci, E; Падуя, Л. (апрель 2013 г.). «Равновесие и участие в ходьбе при фациоскапуло-плечевой дистрофии: пилотное исследование эффектов индивидуальных ортезов нижних конечностей». Европейский журнал физической и реабилитационной медицины. 49 (2): 169–78. PMID 23138679.
- ^ а б c Tawil, R; van der Maarel, S; Падберг, GW; ван Энгелен, Б.Г. (июль 2010 г.). «171-й международный семинар ENMC: Стандарты лечения и лечения фасциально-плечевой мышечной дистрофии». Нервно-мышечные расстройства. 20 (7): 471–5. Дои:10.1016 / j.nmd.2010.04.007. PMID 20554202. S2CID 18448196.
- ^ Sansone, V; Бойнтон, Дж; Паленски, К. (июнь 1997 г.). «Использование золотых гирь для коррекции лагофтальма при нервно-мышечных заболеваниях». Неврология. 48 (6): 1500–3. Дои:10.1212 / wnl.48.6.1500. HDL:2434/210652. PMID 9191754. S2CID 16251273.
- ^ Мацумото, М; Онода, S; Uehara, H; Миура, Y; Катаяма, Й; Кимата, Y (сентябрь 2016 г.). «Коррекция нижней губы с помощью трансплантата хряща и резекции губы у пациентов с лицево-плечевой мышечной дистрофией». Журнал черепно-лицевой хирургии. 27 (6): 1427–9. Дои:10.1097 / SCS.0000000000002720. PMID 27300465. S2CID 16343571.
- ^ Кришнамурти, S; Ибрагим, М. (январь 2019 г.). «Перенос сухожилий при опущении стопы». Индийский журнал пластической хирургии. 52 (1): 100–108. Дои:10.1055 / с-0039-1688105. ЧВК 6664842. PMID 31456618.
- ^ Чиодо, Крис; Блюман, Эрик М. (21.10.2011). Перенос сухожилий в стопе и лодыжке. Сондерс. п. 421. ISBN 9781455709243. Получено 1 января 2020.
- ^ Демирхан, Мехмет; Уйсал, Озгур; Аталар, Ата Джан; Киликоглу, Ондер; Сердароглу, Пирайе (31 марта 2009 г.). «Лопаточно-грудной артродез при лицево-лопаточно-плечевой дистрофии с помощью мультифиламентного кабеля». Клиническая ортопедия и смежные исследования. 467 (8): 2090–2097. Дои:10.1007 / s11999-009-0815-9. ЧВК 2706357. PMID 19333668.
- ^ ДеФранко, Майкл Дж .; Нхо, Шейн; Ромео, Энтони А. (апрель 2010 г.). «Лопаточно-грудной слияние». Журнал Американской академии хирургов-ортопедов. 18 (4): 236–42. Дои:10.5435/00124635-201004000-00006. PMID 20357232. S2CID 27456684.
- ^ Abrams, Jeffrey S .; Белл, Роберт Х .; Токиш, Джон М. (2018). РАСШИРЕННАЯ РЕКОНСТРУКЦИЯ ПЛЕЧЕЙ. АМЕРИКАНСКИЙ АКАД ОРТОПЕДОВ. ISBN 9781975123475.
- ^ а б c Динен Дж.К., Арнс Х., ван дер Маарель С.М., Падберг Г.В., Вершуурен Дж.Дж., Баккер Э., Вайнрайх СС, Вербек А.Л., ван Энгелен Б.Г. (2014). «Заболеваемость и распространенность фациоскапуло-плечевой дистрофии среди населения». Неврология. 83 (12): 1056–9. Дои:10.1212 / WNL.0000000000000797. ЧВК 4166358. PMID 25122204.
- ^ Динен, JC; Horlings, CG; Verschuuren, JJ; Verbeek, AL; ван Энгелен, Б.Г. (2015). "Эпидемиология нервно-мышечных заболеваний: всесторонний обзор литературы". Журнал нервно-мышечных заболеваний. 2 (1): 73–85. Дои:10.3233 / JND-140045. PMID 28198707.
- ^ Теверони, Э; Пеллегрино, М; Sacconi, S; Calandra, P; Cascino, I; Farioli-Vecchioli, S; Пума, А; Гарибальди, М; Morosetti, R; Таска, G; Ricci, E; Тревизан, CP; Галлуцци, G; Понтекорви, А; Крещенци, М; Deidda, G; Моретти, Ф (3 апреля 2017 г.). «Эстрогены усиливают дифференцировку миобластов при фасциально-лопаточно-плечевой мышечной дистрофии за счет противодействия активности DUX4». Журнал клинических исследований. 127 (4): 1531–1545. Дои:10.1172 / JCI89401. ЧВК 5373881. PMID 28263188.
- ^ Мул, К; Horlings, CGC; Воерманс, Северная Каролина; Schreuder, THA; ван Энгелен, BGM (июнь 2018 г.). «Пожизненное воздействие эндогенных эстрогенов и тяжесть заболевания у пациенток с фациоскапуло-плечевой мышечной дистрофией». Нервно-мышечные расстройства. 28 (6): 508–511. Дои:10.1016 / j.nmd.2018.02.012. PMID 29655530.
- ^ Дюшенн, Гийом-Бенджамин (1868). "De la paralysie musculaire pseudo-hypertrophique, ou paralysie myo-sclérosique". Arch. Gen. Med. (На французском). Национальная библиотека Франции. 11: 5–25, 179–209, 305–321, 421–443, 552–588. Получено 18 мая 2020.
- ^ а б Синдром Ландузи-Дежерина, whonamedit.com, дата обращения 11 марта 2007 г.
- ^ Ландузи; Дежерин (1884). «Прогрессирующая атрофическая миопатия (myopathie héréditaire, débutant dans l'enfance par la face, sans altération du système nerveux)». Comptes Rendus de l'Académie des Sciences. 98: 53–55.
- ^ Ландузи; Дежерин (1886). «Вклад в исследование прогрессирующей атрофической миопатии (прогрессирующая атрофическая миопатия, скапуло-гумеральная типа)». Comptes Rendus des Séances de la Société de Biologie. 38: 478–481.
- ^ Тайлер, Фрэнк; Стивенс Ф. Э. (апрель 1950 г.). «Исследования при мышечных нарушениях. II. Клинические проявления и наследование фасциально-плечевой дистрофии в многодетной семье». Анналы внутренней медицины. 32 (4): 640–660. Дои:10.7326/0003-4819-32-4-640. PMID 15411118.
- ^ Кениг, М; Хоффман, EP; Бертельсон, CJ; Монако, АП; Feener, C; Kunkel, LM (31 июля 1987 г.). «Полное клонирование кДНК мышечной дистрофии Дюшенна (МДД) и предварительная геномная организация гена МДД у здоровых и больных людей». Клетка. 50 (3): 509–517. Дои:10.1016/0092-8674(87)90504-6. PMID 3607877. S2CID 35668717.
- ^ Wijmenga, C; Падберг, GW; Moerer, P; и другие. (Апрель 1991 г.). «Картирование гена facioscapulohumeral мышечной дистрофии к хромосоме 4q35-qter с помощью анализа многоточечного сцепления и гибридизации in situ». Геномика. 9 (4): 570–575. Дои:10.1016 / 0888-7543 (91) 90348-И. PMID 2037288.
- ^ Гилберт-младший; Stajich, JM; Стена, S; и другие. (Август 1993 г.). «Доказательства гетерогенности при фасциоскапуло-плечевой мышечной дистрофии (ЛЛД)». Американский журнал генетики человека. 53 (2): 401–408. ЧВК 1682358. PMID 8328457.
- ^ а б Винокур, СТ; Bengtsson, U; Feddersen, J; и другие. (Май 1994). «Перестройка ДНК, связанная с фациоскапуло-плечевой мышечной дистрофией, включает повторяющийся элемент, связанный с гетерохроматином: последствия для роли структуры хроматина в патогенезе заболевания». Хромосомные исследования. 2 (3): 225–234. Дои:10.1007 / bf01553323. PMID 8069466. S2CID 6933736.
- ^ Hewitt, JE; Лайл, Р; Кларк, LN; и другие. (Август 1994 г.). «Анализ локуса тандемных повторов D4Z4, ассоциированного с фациоскапуло-плечевой мышечной дистрофией». Молекулярная генетика человека. 3 (8): 1287–1295. Дои:10,1093 / hmg / 3.8.1287. PMID 7987304.
- ^ Гилберт-младший; Speer, MC; Stajich, J; и другие. (Октябрь 1995 г.). «Картирование исключения хромосомных областей, которые перекрестно гибридизуются с маркерами, ассоциированными с FSHD1A в FSHD1B». Журнал медицинской генетики. 32 (10): 770–773. Дои:10.1136 / jmg.32.10.770. ЧВК 1051697. PMID 8558552.
- ^ van Deutekom, JC; Lemmers, RJ; Grewal, PK; и другие. (Май 1996 г.). «Идентификация первого гена (FRG1) из области FSHD на хромосоме 4q35 человека». Молекулярная генетика человека. 5 (5): 581–590. Дои:10,1093 / чмг / 5.5.581. PMID 8733123.
- ^ а б Kowaljow, V; Marcowycz, A; Анссо, Э; и другие. (Август 2007 г.). «Ген DUX4 в локусе FSHD1A кодирует проапоптотический белок». Нервно-мышечные расстройства. 17 (8): 611–623. Дои:10.1016 / j.nmd.2007.04.002. PMID 17588759. S2CID 25926418.
- ^ Габриэлс, Дж; Beckers, MC; Дин, Н; и другие. (5 августа 1999 г.). «Нуклеотидная последовательность частично удаленного локуса D4Z4 у пациента с ЛЛД идентифицирует предполагаемый ген в каждом элементе размером 3,3 т.п.н.». Ген. 236 (1): 25–32. Дои:10.1016 / S0378-1119 (99) 00267-X. PMID 10433963.
- ^ Цзянь, Ф; Вс, В; Хопкинс, NE; и другие. (Ноя 2001). «Метилирование субтеломерного повтора, связанного с синдромом FSHD, в культурах и тканях нормальных и FSHD клеток». Молекулярная генетика и метаболизм. 74 (3): 322–331. Дои:10.1006 / mgme.2001.3219. PMID 11708861.
- ^ Lemmers, RJ; де Киевит, П; Sandkuijl, L; и другие. (Октябрь 2002 г.). «Фациоскапуло-плечевая мышечная дистрофия однозначно связана с одним из двух вариантов субтеломера 4q». Природа Генетика. 32 (2): 235–236. Дои:10,1038 / ng999. PMID 12355084. S2CID 28107557.
- ^ Габеллини, Д; Грин, MR; Tupler, R (9 августа 2002 г.). «Несоответствующая активация гена при ЛЛД: репрессорный комплекс связывает хромосомный повтор, удаленный в дистрофической мышце». Клетка. 110 (3): 339–348. Дои:10.1016 / S0092-8674 (02) 00826-7. PMID 12176321. S2CID 16396883.
- ^ ван Овервельд, PG; Lemmers, RJ; Sandkuijl, LA; и другие. (Декабрь 2003 г.). «Гипометилирование D4Z4 при 4q-связанной и не-4q-связанной фасциоскапуло-плечевой мышечной дистрофии». Природа Генетика. 35 (4): 315–317. Дои:10,1038 / ng1262. PMID 14634647. S2CID 28696708.
- ^ Lemmers, RJ; Вольгемут, М; Франц, Р.Р .; Падберг, GW; Morava, E; ван дер Маарель, С.М. (декабрь 2004 г.). «Сокращения D4Z4 на субтеломерах 4qB не вызывают фациоскапуло-плечевую мышечную дистрофию». Американский журнал генетики человека. 75 (6): 1124–1130. Дои:10.1086/426035. ЧВК 1182148. PMID 15467981.
- ^ Габеллини, Д; Д'Антона, G; Moggio, M; и другие. (23 февраля 2006 г.). «Facioscapulohumeral мышечная дистрофия у мышей с избыточной экспрессией FRG1». Природа. 439 (7079): 973–977. Bibcode:2006Натура 439..973Г. Дои:10.1038 / природа04422. PMID 16341202. S2CID 4427465.
- ^ Клапп, Дж; Митчелл, Л. М.; Болланд, диджей; и другие. (Август 2007 г.). «Эволюционное сохранение кодирующей функции для D4Z4, тандемного повтора ДНК, мутировавшего при фациоскапуло-плечевой мышечной дистрофии». Американский журнал генетики человека. 81 (2): 264–279. Дои:10.1086/519311. ЧВК 1950813. PMID 17668377.
- ^ Диксит, М; Анссо, Э; Тассин, А; и другие. (13 ноября, 2007). «DUX4, ген-кандидат фациоскапуло-плечевой мышечной дистрофии, кодирует активатор транскрипции PITX1». Труды Национальной академии наук США. 104 (46): 18157–18162. Bibcode:2007PNAS..10418157D. Дои:10.1073 / pnas.0708659104. ЧВК 2084313. PMID 17984056.
- ^ de Greef, JC; Lemmers, RJ; van Engelen, BG; и другие. (Октябрь 2009 г.). «Общие эпигенетические изменения D4Z4 при сокращении зависимой и независимой от сокращения ЛЛД». Человеческая мутация. 30 (10): 1449–1459. CiteSeerX 10.1.1.325.8388. Дои:10.1002 / humu.21091. PMID 19728363. S2CID 14517505.
- ^ Снайдер, L; Asawachaicharn, A; Тайлер, AE; и другие. (1 июля 2009 г.). «РНК-транскрипты, фрагменты размером с миРНК и белки, полученные из единиц D4Z4: новые кандидаты в патофизиологию фациоскапуло-плечевой дистрофии». Молекулярная генетика человека. 18 (13): 2414–2430. Дои:10,1093 / hmg / ddp180. ЧВК 2694690. PMID 19359275.
- ^ а б Колата, Джина (19 августа 2010 г.). «Было обнаружено, что реанимированная« мусорная »ДНК вызывает болезнь». Нью-Йорк Таймс. Получено 29 августа 2010.
- ^ Scionti, I; Греко, F; Ricci, G; и другие. (6 апреля 2012 г.). «Масштабный популяционный анализ ставит под сомнение современные критерии молекулярной диагностики фасциоскапуло-плечевой мышечной дистрофии». Американский журнал генетики человека. 90 (4): 628–635. Дои:10.1016 / j.ajhg.2012.02.019. ЧВК 3322229. PMID 22482803.
- ^ Geng, LN; Yao, Z; Снайдер, L; и другие. (17 января 2012 г.). «DUX4 активирует гены зародышевой линии, ретроэлементы и иммунные медиаторы: последствия для фациоскапуло-плечевой дистрофии». Клетка развития. 22 (1): 38–51. Дои:10.1016 / j.devcel.2011.11.013. ЧВК 3264808. PMID 22209328.
- ^ Джонс, Т.И.; Chen, JC; Рагимов, Ф; и другие. (15 октября 2012 г.). «Семейные исследования лицево-капуло-плечевой мышечной дистрофии экспрессии DUX4: данные о модификаторах заболевания и количественная модель патогенеза». Молекулярная генетика человека. 21 (20): 4419–4430. Дои:10.1093 / hmg / dds284. ЧВК 3459465. PMID 22798623.
- ^ Кром, Ю.Д .; Thijssen, PE; Янг, JM; и другие. (Апрель 2013 г.). «Внутренняя эпигенетическая регуляция макросателлитного повтора D4Z4 в модели трансгенных мышей для FSHD». PLOS Genetics. 9 (4): e1003415. Дои:10.1371 / journal.pgen.1003415. ЧВК 3616921. PMID 23593020.
- ^ Ferreboeuf, M; Марио, V; Bessieres, B; и другие. (1 января 2014 г.). «Последующие гены-мишени DUX4 и DUX4 экспрессируются в мышцах FSHD плода». Молекулярная генетика человека. 23 (1): 171–181. Дои:10.1093 / hmg / ddt409. PMID 23966205.
- ^ а б Коэн, Джастин; Дезимоун, Алек; Лек, Монкол; Лек, Анджела (октябрь 2020 г.). «Терапевтические подходы при лицево-лопаточно-плечевой мышечной дистрофии». Тенденции в молекулярной медицине: S1471491420302392. Дои:10.1016 / j.molmed.2020.09.008. PMID 33092966.
- ^ Tawil, R; Макдермотт, член парламента; Пандья, S; Король, W; Кисель, Дж; Mendell, JR; Григгс, Р.К. (январь 1997 г.). «Пилотное испытание преднизона при фациоскапуло-плечевой мышечной дистрофии. Группа FSH-DY». Неврология. 48 (1): 46–9. Дои:10.1212 / wnl.48.1.46. PMID 9008492. S2CID 729275.
- ^ Кисель, JT; Макдермотт, член парламента; Натараджан, Р. Mendell, JR; Пандья, S; Король, WM; Григгс, Р. Тавил, Р. (май 1998 г.). «Пилотное испытание альбутерола при фациоскапуло-плечевой мышечной дистрофии. Группа FSH-DY». Неврология. 50 (5): 1402–6. Дои:10.1212 / wnl.50.5.1402. PMID 9595995. S2CID 24848310.
- ^ Кисель, JT; Макдермотт, член парламента; Mendell, JR; Король, WM; Пандья, S; Григгс, Р. Tawil, R; ФСХ-ДЙ, Группа. (23 октября 2001 г.). «Рандомизированное двойное слепое плацебо-контролируемое исследование альбутерола при фациоскапуло-плечевой дистрофии». Неврология. 57 (8): 1434–40. Дои:10.1212 / wnl.57.8.1434. PMID 11673585. S2CID 28093111.
- ^ van der Kooi, EL; Vogels, OJ; ван Асселдонк, Р.Дж.; Lindeman, E; Хендрикс, JC; Вольгемут, М; van der Maarel, SM; Падберг, GW (24 августа 2004 г.). «Силовые тренировки и альбутерол при фасциально-плечевой мышечной дистрофии». Неврология. 63 (4): 702–8. Дои:10.1212 / 01.wnl.0000134660.30793.1f. PMID 15326246. S2CID 22778327.
- ^ а б Кэмпбелл, AE; Oliva, J; Йейтс, депутат; Чжун, JW; Шадл, Южная Каролина; Снайдер, L; Singh, N; Тай, S; Хирамуки, Y; Tawil, R; van der Maarel, SM; Tapscott, SJ; Свердруп, FM (4 сентября 2017 г.). «Ингибиторы бромодомена BET и агонисты бета-2-адренергического рецептора, идентифицированные в результате скрининга соединений, которые ингибируют экспрессию DUX4 в мышечных клетках FSHD». Скелетные мышцы. 7 (1): 16. Дои:10.1186 / s13395-017-0134-х. ЧВК 5584331. PMID 28870238.
- ^ Elsheikh, BH; Bollman, E; Перуджа, М; Король, W; Гэллоуэй, G; Кисель, JT (24 апреля 2007 г.). «Экспериментальное испытание дилтиазема при фациально-плечевой мышечной дистрофии». Неврология. 68 (17): 1428–9. Дои:10.1212 / 01.wnl.0000264017.08217.39. PMID 17452589. S2CID 361422.
- ^ Wyeth инициирует клинические испытания с помощью исследуемой терапии мышечной дистрофии MYO-029
- ^ Малькольм, Эмили (18 июня 2019 г.). «АСЕ-083». Новости мышечной дистрофии. Получено 19 декабря 2019.
- ^ Киношита, июнь. «Что в имени? - Общество ЛЛДП». Общество FSHD. Получено 18 августа 2019.
- ^ "Общество ФСХ".
- ^ «FSHD Europe: 光 の 中 の た こ 焼 き を 食 べ た い と 思 っ て 30 年 - な ん の こ っ ち ゃ!».
- ^ "Fulcrum Therapeutics приобретает глобальные права на лосьмапимод, потенциально модифицирующий заболевание препарат для лечения лицево-капуло-плечевой мышечной дистрофии". BioSpace. Получено 12 августа 2019.
- ^ «Эффективность и безопасность лосмапимода у субъектов с лицево-капуло-плечевой мышечной дистрофией (ЛЛД)». ClinicalTrials.gov. Национальная медицинская библиотека США. Получено 12 августа 2019.
- ^ DeSimone, Alec M .; Лешик, Джон; Вагнер, Кэтрин; Эмерсон, Чарльз П. (11 декабря 2019 г.). «Идентификация пути гиалуроновой кислоты как терапевтической мишени для фациоскапуло-плечевой мышечной дистрофии». Достижения науки. 5 (12): eaaw7099. Bibcode:2019SciA .... 5.7099D. Дои:10.1126 / sciadv.aaw7099. ЧВК 6905861. PMID 31844661.
- ^ Боснаковский, Д; да Силва, MT; Солнечный, ST; Ener, ET; Тосо, EA; Юань, C; Cui, Z; Уолтерс, Массачусетс; Джадхав, А; Киба, М. (сентябрь 2019 г.). «Новый ингибитор P300 обращает DUX4-опосредованное глобальное гиперацетилирование гистона H3, экспрессию целевого гена и гибель клеток». Достижения науки. 5 (9): eaaw7781. Bibcode:2019SciA .... 5.7781B. Дои:10.1126 / sciadv.aaw7781. ЧВК 6739093. PMID 31535023.
- ^ "Facio представить на Конгрессе Всемирного общества мышц". Лицевая терапия. 30 сентября 2019 г.. Получено 9 ноября 2019.
- ^ «Facio раскрывает новый механизм, нацеленный на причину ЛЛПД». Лицевая терапия. 24 июн 2019. Получено 9 ноября 2019.
- ^ Passerieux, E; Хаёт, М; Jaussent, A; Карнак, G; Gouzi, F; Pillard, F; Пико, MC; Böcker, K; Hugon, G; Pincemail, J; Дефрань, Джо; Веррипс, Т; Мерсье, Дж; Лаудж-Шенивесс, Д. (апрель 2015 г.). «Влияние добавок витамина C, витамина E, глюконата цинка и селенометионина на функцию мышц и биомаркеры окислительного стресса у пациентов с фациоскапуло-плечевой дистрофией: двойное слепое рандомизированное контролируемое клиническое исследование» (PDF). Свободная радикальная биология и медицина. 81: 158–69. Дои:10.1016 / j.freeradbiomed.2014.09.014. PMID 25246239.
- ^ Дэни, Антуан; Барб, Корали; Рапин, Амандин; Ревейер, Кристиан; Хардуэн, Жан-Бенуа; Морроне, Изабелла; Волак-Тьерри, Аврора; Драме, Мустафа; Кальмус, Арно; Саккони, Сабрина; Бассез, Гийом; Тиффро, Винсент; Ричард, Изабель; Галле, Бенджамин; Приджент, Элен; Тайар, Редха; Веселый, Дэмиен; Новелла, Жан-Люк; Бойер, Франсуа Констан (2015). «Создание анкеты качества жизни для медленно прогрессирующих нервно-мышечных заболеваний». Исследование качества жизни. 24 (11): 2615–2623. Дои:10.1007 / s11136-015-1013-8. ISSN 0962-9343. PMID 26141500. S2CID 25834947.
внешняя ссылка
- список клинических исследований ЛЛПД, Clinicaltrials.gov.
- fsh в Национальные институты здравоохранения США /UW Генные тесты
| Классификация | |
|---|---|
| Внешние ресурсы |
| Типы | |
|---|---|
| Национальные / международные организации |
|
| Национальные / международные мероприятия |
|
| Клинические испытания | |