Семейный аденоматозный полипоз - Familial adenomatous polyposis
| Семейный аденоматозный полипоз | |
|---|---|
| Другие имена | ФАП |
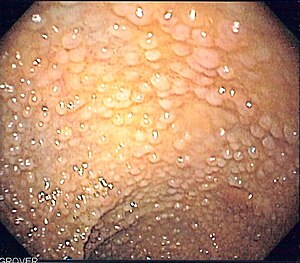 | |
| Эндоскопический изображение сигмовидная кишка пациента с семейным аденоматозным полипозом | |
| Специальность | Гастроэнтерология, Онкология |
| Осложнения | Колоректальный рак |
| Обычное начало | <35 лет |
| Продолжительность | На всю жизнь |
| Типы | Классический или разбавленный |
| Причины | Мутация гена APC |
| Диагностический метод | Колоноскопия Генетическое тестирование |
| Дифференциальная диагностика | Синдром Линча, MUTYH-ассоциированный полипоз |
| Уход | Колоноскопия Полипэктомия Верхняя эндоскопия Колэктомия |
| Частота | 1 из 10 000–15 000 |
Семейный аденоматозный полипоз (ФАП) является аутосомно-доминантный наследственное состояние, при котором многочисленные аденоматозный полипы формируются в основном в эпителий из толстая кишка. Пока эти полипы начинаются доброкачественный, злокачественная трансформация в рак толстой кишки возникает, когда их не лечить. Известно три варианта: FAP и ослабленный ФАП (первоначально назывался синдром наследственной плоской аденомы[1]) вызваны Ген APC дефекты хромосомы 5 в то время как аутосомно-рецессивный ФАП (или же MUTYH-ассоциированный полипоз) вызвано дефектами в МУТЫХ ген на хромосоме 1. Самым тяжелым и распространенным из этих трех является САП; хотя для всех трех образующиеся полипы толстой кишки и рак первоначально ограничиваются стенкой толстой кишки. Обнаружение и удаление до метастаз вне толстой кишки может значительно уменьшить и во многих случаях устранить распространение рака.
Подразумевается, что первопричиной FAP является генетическая мутация - изменение в организме гены-супрессоры опухолей предотвращающие развитие опухолей. Это изменение позволяет многочисленным клеткам стенки кишечника развиваться в потенциально раковые полипы, когда они обычно достигают конца своей жизни; неизбежно один или несколько в конечном итоге будут развиваться и приведут к рак (Риск 7% к 21 году, возрастает до 87% к 45 годам и 93% к 50 годам). Эти изменения генов не вызывают рак, а, скорее, снижают способность организма предотвращать превращение клеток в раковые. Даже с изменением гена может пройти время, прежде чем действительно разовьется клетка, которая в результате станет злокачественной, и в некоторых случаях ген может все еще частично работать, чтобы контролировать опухоли, поэтому для развития рака от FAP требуется много лет, и он почти всегда болезнь, начинающаяся у взрослых.
Вторая форма ФАП, известная как ослабленный семейный аденоматозный полипоз имеет функциональный, но слегка нарушенный ген APC. Таким образом, он может работать как обычно. Ослабленный FAP по-прежнему представляет собой высокий 70% -ный риск развития рака в течение жизни (по оценкам), но обычно проявляется гораздо меньшим количеством полипов (обычно 30), чем сотни или тысячи, обычно встречающиеся при FAP.[2] и возникает в возрасте, когда FAP обычно больше не считается вероятным - обычно между 40 и 70 годами (в среднем 55[3]), а не более обычные 30-е и выше. Поскольку у него гораздо меньше полипов, варианты лечения могут быть разными.[2]
Третий вариант, аутосомно-рецессивный семейный аденоматозный полипоз или же MUTYH-ассоциированный полипоз, также мягче и, как следует из названия, требует оба родителя должны быть "носителями" проявить состояние.
В некоторых случаях FAP может проявляться в толстой кишке выше, чем обычно (например, восходящая кишка,[нужна цитата ] или рядом с селезеночный изгиб, или в желудке или двенадцатиперстной кишке[1]), где они не проявляют никаких симптомов до тех пор, пока рак не появится и не сильно продвинется. Мутации APC были связаны с некоторыми другими видами рака, такими как рак щитовидной железы. Поскольку мутация, вызывающая FAP, является аутосомно-доминантной, она может быть унаследована напрямую от любого родителя ребенку. А генетический анализ крови гена APC, который может определить, присутствует ли он, и, следовательно, может предсказать возможность FAP. Людям из группы риска (из-за семейных связей или генетического тестирования) обычно предлагается рутинный мониторинг кишечного тракта каждые 1-3 года в течение всей жизни, начиная с полового созревания для FAP и в раннем взрослом возрасте для аттенуированных форм. Операция по резекции толстой кишки рекомендуется, если обнаружены многочисленные полипы толстой кишки из-за высокого риска ранней смерти от рака толстой кишки. Существуют международные регистры полипоза, которые отслеживают известные случаи дефектов генов FAP или APC для исследовательских и клинических целей. Мутация APC также часто встречается в отдельных случаях колоректальной карциномы, что подчеркивает ее важность при этой форме рака.
Признаки и симптомы
С раннего подросткового возраста у пациентов с этим заболеванием постепенно (и в большинстве случаев бессимптомно) развиваются от сотен до тысяч колоректальные полипы (и иногда полипы в других местах ) - мелкие аномалии на поверхности кишечного тракта, особенно в толстая кишка в том числе двоеточие или же прямая кишка. Они могут кровоточить, что приводит к появлению крови в стуле. Если кровь не видна, у пациента все еще возможно развитие анемия из-за постепенно развивающегося дефицита железа. Если злокачественное новообразование развивается, это может проявляться потеря веса, изменение привычки кишечника, или даже метастаз к печень или где-нибудь еще. ФАП также может развиваться «незаметно» у некоторых людей, подавая мало признаков или не проявляя никаких признаков до тех пор, пока не перерастет в прогрессирующую. колоректальный рак.
Поскольку семейный полипоз развивается очень постепенно с годами, а также может проявляться в «ослабленной» форме еще более постепенно, полипы, возникающие в результате FAP, могут привести к развитию рака в любой момент от подросткового до пожилого возраста.
В зависимости от природы дефекта в гене APC и от того, является ли он полной или ослабленной формой, семейный полипоз может проявляться в виде полипов в толстой или толстой кишке. дуоденальный тракт, или в любой их комбинации. Следовательно, отсутствие полипов, например, в прямой кишке, само по себе может быть недостаточным для подтверждения отсутствия полипов. Возможно, потребуется рассмотреть и визуально осмотреть другие возможные части кишечного тракта. Колоноскопия предпочтительнее ректороманоскопия для этого, поскольку он обеспечивает лучшее наблюдение за обычным расположением полипов справа.[1]

Генетическая детерминанта семейного полипоза может также предрасполагать носителей к другим злокачественным новообразованиям, например, двенадцатиперстной кишки и желудок (особенно ампулярная аденокарцинома). Другими признаками, которые могут указывать на ФАП, являются пигментные поражения сетчатка («CHRPE - врожденная гипертрофия пигментного эпителия сетчатки»), кисты челюсти, сальные кисты, и остеомы (доброкачественные опухоли костей). Сочетание полипоза, остеомы, фибромы и сальные кисты Называется Синдром Гарднера (с аномальными рубцами или без них).[4]
Генетика
Семейный аденоматозный полипоз может иметь разные модели наследования и разные генетические причины. Когда это состояние является результатом мутаций в APC ген, наследуется в аутосомно-доминантный паттерн, что означает, что одной копии измененного гена достаточно, чтобы вызвать нарушение. Заболеваемость злокачественными новообразованиями в этих случаях приближается к 100%. В большинстве случаев у больного есть один родитель с этим заболеванием.
Варианты мутации гена APC
В APC это ген-супрессор опухоли отвечает за производство аденоматозный полипоз кишечной палочки (APC), большой многофункциональный белок, подавляющий опухоль, который действует как «привратник», предотвращающий развитие опухолей. (APC регулирует β-катенин, белок, который играет решающую роль в клеточной коммуникации, передаче сигналов, росте и контролируемом разрушении, но который также остается неконтролируемым вызывает многочисленные виды рака[1]). Недостаток в гене APC означает, что APC не так эффективен, как должен быть, и со временем вполне вероятно, что некоторые клетки, которые должны были контролироваться APC, перестанут действовать, а вместо этого будут продолжать развиваться и становиться злокачественными. При знакомом полипозе они обычно проявляются как полипы - небольшие аномалии на поверхности кишечного тракта.
Хотя полипы по своей природе доброкачественные, первый шаг гипотеза с двумя ударами уже произошло: унаследованная мутация APC. Часто оставшиеся «нормальные» аллель мутирует или удаляется, что ускоряет образование полипов. Дальнейшие мутации (например, в p53 или же kRAS) к APC-мутированным клеткам с гораздо большей вероятностью могут привести к раку, чем к немутантным эпителиальный клетки.
Нормальное функционирование продукта гена APC все еще исследуется; присутствует как ядро клетки и мембрана. Каноническая опухолевая супрессорная функция APC заключается в подавлении β-катенина, но другие опухолевые супрессорные функции APC могут быть связаны с адгезией клеток и цитоскелет организация.
Мутация APC также часто встречается в отдельных случаях колоректального рака, что подчеркивает его важность при этой форме рака.
Варианты мутации гена MUTYH
МУТЫХ кодирует Ремонт ДНК фермент MYH гликозилаза. Во время нормальной клеточной деятельности гуанин иногда изменяется кислород, что заставляет его соединяться с аденин вместо цитозин. Гликозилаза MYH исправляет эти ошибки с помощью базовая эксцизионная пластика, так что мутации не накапливаются в ДНК и приводят к образованию опухоли. Когда гликозилаза MYH не функционирует должным образом, могут возникать ошибки ДНК, чтобы инициировать онкогенез с клиническими проявлениями, аналогичными таковым у пациентов с мутациями APC.
Мутации в МУТЫХ гены наследуются в аутосомно-рецессивный Это означает, что две копии гена должны быть изменены, чтобы человек пострадал от заболевания. Чаще всего родители ребенка с аутосомно-рецессивным заболеванием не страдают, но являются носителями одной копии измененного гена.
Модели животных
"ApcМин."модель мыши была описана в 1990 году и имеет Apc аллель со стоп-кодоном в положении 850. Гетерозиготность этой мутации приводит к полному пенетрантному фенотипу на большинстве генетических фонов, при этом у мышей на чувствительном фоне развивается более 100 опухолей в кишечном тракте. Количество и расположение опухолей кишечника изменяются несвязанными генами. С тех пор появилось много других моделей, в том числе модель с ослабленным FAP (модель 1638N) и несколько условные мутанты которые позволяют тканеспецифичному или временному устранению функции гена. Для получения дополнительной информации см. мышиные модели колоректального и кишечного рака.
В 2007 году модель крысы «ApcPirc» была выделена со стоп-кодоном в положении 1137.[5] В отличие от моделей на мышах, где> 90% опухолей формируются в тонком кишечнике, крысы Pirc образуют опухоли преимущественно (> 60%) в толстом кишечнике, аналогично клиническим проявлениям у человека.
Диагностика

Диагноз FAP до развития рака толстой кишки важен не только для человека, но и для других членов семьи, которые могут быть затронуты. Существуют два метода диагностики:
- Колоноскопия является обычным диагностическим тестом выбора, так как он способствует более распространенному правостороннему расположению полипов, чем ректороманоскопия, если мутация ослаблена FAP,[1] и может подтвердить или разрешить (а) фактическую клиническую картину и любое изменение состояния индивидуума из группы риска, (б) количественную оценку полипов по всей толстой кишке, (в) а гистологический диагноз (определение типа клеток / рака) и (d) при наличии полипов он может подсказать, целесообразно ли амбулаторное иссечение (удаление) пациента или рекомендуется хирургическое вмешательство. Клизма бария и виртуальная колоноскопия (форма медицинской визуализации) также может использоваться, чтобы предложить диагноз ФАП.
- Генетическое тестирование дает окончательный диагноз в 95% случаев; генетическое консультирование обычно требуется в семьях, где был диагностирован ФАП. Тестирование также может помочь в диагностике пограничных случаев в семьях, которые иначе известны как p34.3 и p32.1 (1p34.3 – p32.1). Тестирование может только показать, подвержен ли человек FAP или исключить его (т.е. унаследовал ли он дефектный ген APC). Он не может определить реальное состояние пациента; это можно обнаружить только при непосредственном физическом осмотре.
NCBI заявляет, что врачи должны убедиться, что они понимают «риски, преимущества и ограничения» любого проведенного генетического теста, поскольку в 1997 году «почти для одной трети людей, прошедших оценку на FAP, врач неверно истолковал результаты теста».[6]
После постановки диагноза FAP закройте колоноскопический наблюдение с полипэктомия необходимо.
Пренатальное тестирование возможно, если у пораженного члена семьи выявлена вызывающая заболевание мутация; тем не менее, пренатальное тестирование для выявления расстройств, начинающихся обычно у взрослых, встречается редко и требует тщательного генетическое консультирование.
УЗИ живота и анализы крови оценка функция печени часто выполняются, чтобы исключить метастаз в печень.
Управление

Из-за того, как развивается семейный полипоз, можно иметь генетическое заболевание и, следовательно, подвергаться риску, но пока у него нет полипов или проблем. Следовательно, человеку может быть поставлен диагноз "риск" FAP, и ему может потребоваться регулярное наблюдение, но он (пока) не имеет FAP (то есть несет дефектный ген, но, по-видимому, еще не имеет никаких реальных медицинских проблем в результате этого. ). Клиническое лечение может охватывать несколько областей:
- Выявление лиц, которые могут подвергаться риску FAP: обычно на основании семейного медицинского анамнеза или генетического тестирования
- Диагностика (подтверждение наличия FAP) - это может быть сделано либо путем генетического тестирования, которое является окончательным, либо путем визуального осмотра самого кишечного тракта.
- Важно отметить, что визуальный осмотр или наблюдение не может «очистить» человека от риска. Он может только сказать, в каком они состоянии сейчас. Если в какой-то момент жизни у человека разовьются многочисленные полипы, это может указывать на диагноз ФАП. (Отсутствие полипов не «очищает» человека, поскольку полипы могут развиться в более позднем возрасте; также несколько полипов с течением времени не так уж редки у людей без ФАП. существенный номер или изобилие полипов обычно указывает на диагноз ФАП, и гистопатология чтобы определить, являются ли полипы злокачественными.)
- Программы скрининга / мониторинга включают визуальный осмотр кишечного тракта для проверки его здорового состояния. Это проводится в обычном порядке каждые несколько лет, если есть основания для беспокойства, когда либо (а) генетический тест подтвердил риск, либо (б) генетический тест не проводился по какой-либо причине, поэтому фактический риск неизвестен. Скрининг и мониторинг позволяют визуально обнаружить полипоз до того, как он станет опасным для жизни.
- Лечение, обычно какое-либо хирургическое вмешательство, применяется, если полипоз привел к образованию большого количества полипов, значительному риску рака или фактическому раку.
История семьи
NCBI заявляет, что «Хотя у большинства людей с диагнозом APC-ассоциированного полипоза есть пораженный родитель, семейный анамнез может оказаться отрицательным из-за неспособности распознать заболевание у членов семьи, ранней смерти родителя до появления симптомов, или позднее начало заболевания у пораженного родителя ».[2] Кроме того, около 20% случаев являются de novo мутации, и из тех, у кого есть очевидная мутация APC de novo (то есть неизвестный семейный анамнез), 20% имеют соматический мозаицизм.[7] Также известно, что существуют бессимптомные люди (и, следовательно, бессимптомные члены семьи).[2]
Мониторинг
Мониторинг предполагает предоставление амбулаторный колоноскопия, а иногда и верхних отделов желудочного тракта эзофагогастродуоденоскопия (EGD, для поиска предраковых заболеваний желудка или рак двенадцатиперстной кишки[1]), как правило, один раз в 1-3 года, и / или генетический анализ крови для окончательного подтверждения или опровержения предрасположенности. Небольшое количество полипов часто бывает вырезанный (удаляется) во время процедуры, если обнаружено, но если есть более серьезные признаки или цифры, в стационаре хирургия может потребоваться.
NCBI заявляет, что когда у человека идентифицирован FAP или мутации, приводящие к FAP: «Уместно оценить родителей пострадавшего человека (a) с помощью молекулярно-генетического тестирования APC, если вызывающая болезнь мутация известна в пробанд [лицо, впервые идентифицированное с заболеванием] или (б) для клинических проявлений состояния полипоза, связанного с APC ".[2]
Уход
Лечение FAP зависит от генотип. У большинства людей с мутацией APC рак толстой кишки разовьется к 40 годам, хотя менее распространенная аттенуированная версия обычно проявляется в более позднем возрасте (40–70). Соответственно, во многих случаях профилактический хирургическое вмешательство может быть рекомендовано до достижения 25-летнего возраста или после обнаружения при активном наблюдении. Есть несколько хирургических вариантов, которые включают удаление толстой или прямой и толстой кишки.
- Вовлеченная прямая кишка: прямая кишка и часть или вся толстая кишка удаляются. Пациенту может потребоваться илеостомия (постоянный стома где стул уходит в мешок на животе) или подвздошно-анальный мешок реконструкция. Решение об удалении прямой кишки зависит от количества полипов в прямой кишке, а также от семейного анамнеза. Если в прямой кишке мало полипов, толстая кишка частично или полностью удаляется, а тонкая кишка (подвздошная кишка) может быть напрямую соединена с прямой кишкой (илеоректальный анастомоз ).
- Прямая кишка не задействована: часть толстой кишки с полипами может быть удалена, а концы соединены заново (частичная колэктомия ), операция, которая требует значительного времени на заживление, но в значительной степени не влияет на качество жизни.
Профилактическое колэктомия показан при наличии более ста полипов, при наличии полипов с тяжелой дисплазией или при наличии нескольких полипов размером более 1 см.
Лечение двух более легких форм FAP может существенно отличаться от более обычного варианта, так как количество полипов намного меньше, что дает больше возможностей.
В настоящее время исследуются различные препараты для замедления злокачественного перерождения полипов, в первую очередь нестероидные противовоспалительные препараты (НПВП). Было показано, что НПВП значительно уменьшают количество полипов, но обычно не влияют на лечение, поскольку еще слишком много полипов, за которыми необходимо наблюдать и лечить эндоскопически.
Прогноз
До достижения поздних стадий колоректального рака полипы ограничиваются внутренней стенкой и толщиной кишечного тракта и не метастазируют и не «распространяются». Таким образом, при условии, что FAP выявляется и контролируется либо на предраковой стадии, либо когда какие-либо раковые полипы все еще находятся внутри кишечного тракта, операция имеет очень высокий уровень успеха в предотвращении или удалении рака без рецидива, поскольку места, вызывающие рак физически удалены в целом хирургическим вмешательством.
После операции, если частичный колэктомия После этого необходимо колоноскопическое наблюдение за оставшейся ободочной кишкой, поскольку у человека все еще есть риск развития рака толстой кишки. Однако, если бы это произошло, это был бы новый случай полипов, вновь развивающихся в не удаленной части толстой кишки после операции, а не рецидив или метастаз любого рака, удаленного с помощью первоначальной операции.
Эпидемиология
Частота появления мутации составляет от 1 на 10 000 до 1 на 15 000 рождений. К 35 годам у 95% людей с ФАП (> 100 аденом) появляются полипы. Без колэктомии рак толстой кишки практически неизбежен. Средний возраст рака толстой кишки у нелеченных лиц составляет 39 лет (от 34 до 43 лет).
Ослабленный FAP возникает, когда APC неисправен, но все еще работает. В результате он частично сохраняет способность подавлять полипы. Следовательно, аттенуированный FAP проявляется как колоректальный рак необычно поздно (возраст 40–70 лет, в среднем = 55 лет).[3]), и обычно с небольшим или, по крайней мере, гораздо меньшим количеством полипов (обычно 30[2]), чем более обычная версия FAP, в том возрасте, когда FAP больше не считается большой вероятностью или риском согласно обычной эпидемиологии FAP.
Сравнение вариантов FAP
В этой таблице сравниваются различные подтипы FAP:[2][1]
| Элемент | ФАП | Ослабленный ФАП | MUTYH Associated FAP |
| Ген | APC | APC | МУТЫХ |
| Типичное проявление полипа | Сотни / тысячи | Менее 100 (0–470, тип 30), иногда плоская, а не полиповидная морфология и более проксимальнее селезеночного изгиба. В исследовании с участием 120 человек 37% (N = 44) имели <10 полипов; У 3 из этих 44 был колоректальный рак]. Также видны полипы фундального отдела желудка и аденомы двенадцатиперстной кишки. Следовательно, полипы и рак могут проявляться в верхний часть толстой кишки или верхний желудочно-кишечный тракт, а не обычные места | ? |
| Типичные основные диагностические критерии | (a) 100+ полипов и возраст до 40 лет, ИЛИ (b) полипы и FAP у родственника | Еще не решено. (а) отсутствие в семейном анамнезе 100+ полипов до 30 лет ПЛЮС ОДИН ИЗ 10–99 полипов / 100+ полипов и в возрасте от 35 до 40 / колоректальный рак до 60 лет и родственников с множественными аденоматозными полипами, ИЛИ (b) Семейный анамнез От 10 до 99 аденом, диагностированных после 30 лет | ? |
| Возраст, в котором проявляются полипы | 7–36 (тип. 16), впоследствии быстро увеличиваясь | ? | ? |
| Риск колоректального рака (пенетрантность ) и возраст при отсутствии лечения | «неизбежно ... практически 100%»: 7% к 21 году, 87% к 45 годам, 93% к 50 годам. Типичный возраст: 34–43 (средний возраст 39) | «Ниже .. менее известна .. по оценкам, 70% к 80 годам». По данным Sovaria, по состоянию на 1998 год, «средний возраст установления диагноза CRC составляет ~ 58 лет». | ? |
| Изменчивость | Меж- и внутрисемейная фенотипическая изменчивость является обычным явлением. | См. FAP | ? |
| Возможные проявления, не связанные с толстой кишкой | «полипы дна желудка и двенадцатиперстной кишки, остеомы, аномалии зубов, врожденная гипертрофия пигментного эпителия сетчатки (CHRPE), опухоли мягких тканей, десмоидные опухоли и связанные с ними виды рака» | Что касается FAP, но «CHRPE и десмоидные опухоли встречаются редко», а также рак щитовидной железы. | ? |
| Другие пожизненные риски | «Карцинома тонкой кишки [двенадцатиперстной кишки или периампуллы] 4–12% [дистальнее двенадцатиперстной кишки] Редко; Аденокарцинома поджелудочной железы ~ 1%; Папиллярная карцинома щитовидной железы 1-2%; ЦНС [тип. Медуллобластома] <1%; Гепатобластома печени 1,6%; Желчь аденокарцинома протоков Низкая, но увеличенная; аденокарцинома желудка <1% в западных культурах ». | ? | ? |
| Наследование | «наследуется по аутосомно-доминантному типу. Приблизительно у 75% -80% людей с APC-ассоциированным полипозом есть пораженный родитель. Потомство пострадавшего человека подвержено 50% риску наследования мутации, вызывающей заболевание» | То же, что и FAP | Другой - рецессивный (носителями должны быть 2 родителя) |
| Генетический обзор и генетическое обнаружение | «Полное секвенирование генов всех экзонов APC и границ интрон-экзон представляется наиболее точным доступным клиническим тестом. Большинство мутаций APC представляют собой бессмысленные мутации или мутации сдвига рамки считывания, которые вызывают преждевременное усечение белка APC. Вероятность обнаружения мутации APC высока зависит от тяжести полипоза толстой кишки и семейного анамнеза. ◦ Примерно 20% лиц с явной мутацией APC de novo. Маркеры, используемые для анализа сцепления в условиях полипоза, связанного с APC, очень информативны и очень тесно связаны с APC-локус; таким образом, их можно использовать с точностью более 98% в более чем 95% семей с APC-ассоциированным полипозом. Тестирование сцепления невозможно для семей с одним затронутым человеком, ситуация, которая часто возникает, когда у индивидуума есть мутация гена de novo, и у него нет пострадавшего потомства. Если болезнь, вызывающая мутацию APC, не обнаружена, молекулярно-генетическое тестирование MUTYH (см. Дифференциальная диагностика) sh следует рассмотреть ". | «Ожидается, что менее 30% людей с ослабленным фенотипом будут иметь идентифицируемую мутацию APC» (см. также подробности в разделе FAP) | ? |
| Генотип-фенотип [основное условие] | Наиболее частая мутация APC находится в кодоне 1309 и приводит к большому количеству полипов в раннем возрасте (~ 20). Обильный полипоз (в среднем = 5000) с мутациями в кодонах 1250–1464. Большинство частичных и полных делеций APC связаны со 100–2000 аденом толстой кишки, хотя наблюдалось ослабление FAP. Пример типичного возраста начала: от кодона 168 до 1580 (исключая 1309) = 30 лет, 5 'кодона 168 и 3' кодона 1580 = 52 года. | Ослабленный FAP связан с мутациями (обычно с усечением) в 5'-части гена (кодоны 1–177), экзоне 9 и дистальном 3'-конце гена; интерстициальные делеции хромосомы 5q22, включающие APC; частичные и целые делеции гена; и соматический мозаицизм для мутаций APC, которые обычно связаны с классическим FAP. Sovaria заявляет, что аттенуированный FAP «вызван мутациями в трех разных областях гена APC - 5'-конце в области, охватывающей экзоны 4 и 5, экзон 9 и крайний 3'-конец. Фенотипическая экспрессия в этих трех группах родственных связей является переменная, но определенно более мягкая, чем при классическом FAP »и что полипы прямой кишки редки при ослабленном FAP, но еще не подтверждено, означает ли это также, что риск рака прямой кишки также ниже. | ? |
| Генотип – фенотип [Другие состояния вне толстой кишки] | Выраженные экстраколонические проявления часто коррелируют (хотя и не полностью) с более дистальными мутациями APC. Общее исследование ФАП плюс экстраколонические симптомы показало:: мутации в кодонах 1395–1493 имеют значительно более высокую частоту возникновения десмоидных опухолей, остеом и эпидермоидных кист, чем мутации с мутациями в кодонах 177–452; мутации в кодонах 1395–1493 имеют значительно более высокую частоту возникновения десмоидных опухолей и остеом, чем мутации с мутациями в кодонах 457–1309; ни у одного человека с мутациями в кодонах 177–452 не развились остеомы или периампулярный рак; только у людей с мутациями в кодонах 457–1309 развиваются гепатобластома и / или опухоли головного мозга. Аденомы двенадцатиперстной кишки: Четырехкратное увеличение риска при мутациях между кодонами 976 и 1067. Десмоидные опухоли: мутации 3 ’в кодоне 1399 были связаны с развитием десмоидной опухоли с отношением шансов 4,37; десмоидные опухоли у 20% индивидов с мутациями 5 'в кодоне 1444, 49% индивидов с мутациями 3' в кодоне 1444 и 61% индивидов с мутациями в кодонах 1445–1580; несколько семей с тяжелыми десмоидными опухолями имели мутации на крайнем 3'-конце; последовательная ассоциация десмоидных опухолей с мутациями дистальнее кодона 1444. CHRPE связан с: мутациями между кодонами 311 и 1444; целые делеции гена APC. Рак щитовидной железы и ФАП: У 24 человек большинство идентифицированных мутаций относились к кодону 1220 от 5 '[Cetta et al. 2000]; 9 из 12 человек имели мутации APC, идентифицированные проксимальнее области кластера мутаций (кодоны 1286–1513). Общий обзор литературы (до августа 2006 г.): выявлено 89 субмикроскопических делеций APC (42 частичных и 47 целых делеций). Экстраколонические находки наблюдались в 36% случаев, без существенных различий в случаях с частичными и целыми делециями гена. | ? | ? |
| Распространенность | «От 2,29 до 3,2 на 100 000 человек. На полипоз, связанный с APC, исторически приходилось около 0,5% всех случаев рака прямой кишки; этот показатель снижается, поскольку все больше членов семьи из группы риска проходят успешное лечение после раннего обнаружения полипа и профилактической колэктомии». | «Вероятно, диагноз недооценен, учитывая меньшее количество полипов толстой кишки и более низкий риск колоректального рака по сравнению с классическим FAP» | ? |
| Лечение проявлений | Классический FAP: «Колэктомия рекомендуется после появления аденом; колэктомия может быть отложена в зависимости от размера и количества аденоматозных полипов. Колэктомия обычно рекомендуется, когда развилось более 20 или 30 аденом или несколько аденом с расширенной гистологией» | «Колэктомия может быть необходима, но примерно у одной трети людей количество полипов толстой кишки достаточно ограничено, поэтому достаточно наблюдения с периодической колоноскопической полипэктомией» | ? |
| Мероприятия по надзору (мониторингу) после установления риска | «Ригмоидоскопия или колоноскопия каждые 1-2 года, начиная с возраста 10–12 лет; колоноскопия при обнаружении полипов; ежегодная колоноскопия, если колэктомия откладывается более чем через год после появления полипов (возраст от 10 до 20 лет с некоторыми более легкими симптомами, может быть рассмотрена отсрочка колэктомии); эзофагогастродуоденоскопия (EGD) к возрасту 25 лет или до колэктомии и повторяется каждые 1-3 года; в некоторых случаях эндоскопическая ретроградная холангиопанкреатография (ERCP) для оценки аденом общего желчного протока; Визуализация кишечника при обнаружении аденомы двенадцатиперстной кишки или перед колэктомией, повторяется каждые 1-3 года в зависимости от результатов; скрининг на гепатобластому (оптимальный интервал неизвестен, одна статья рекомендует «не реже одного раза в три месяца»); ежегодное физикальное обследование, включая оценку внекишечного проявлений и пальпации щитовидной железы с учетом повторного ультразвукового исследования и тонкоигольной аспирации при наличии узлов щитовидной железы » | "Колоноскопия каждые два-три года, начиная с 18-20 лет; эзофагогастродуоденоскопия (EGD) начинается в возрасте 25 лет или до колэктомии и повторяется каждые 1-3 года; в некоторых случаях может потребоваться эндоскопическая ретроградная холангиопанкреатография (ERCP) для оценки аденом общего желчного протока; ежегодное физикальное обследование с пальпацией щитовидной железы с учетом последующего ультразвукового исследования и тонкоигольной аспирации при наличии узлов щитовидной железы. Колэктомия обычно рекомендуется при наличии более 20 или 30 аденом или множественных аденом с развитой гистологией ». По состоянию на 1998 год Sovaria заявляла, что «колоноскопия, в отличие от ректороманоскопии, должна быть рекомендована для эндоскопического наблюдения из-за правостороннего расположения колоректальных аденом; эндоскопическое наблюдение UGI оправдано в попытке обнаружить предраковые опухоли желудка или двенадцатиперстной кишки; индивидуумы с [ослабленным FAP] может потребоваться полная колэктомия с илео-ректальным анастомозом только в том случае, если рекомендуется профилактическая колэктомия » | ? |
| Решение контролировать | «Раннее распознавание может позволить своевременное вмешательство и улучшить конечный результат; таким образом, уместно наблюдение за бессимптомными детьми из группы риска на предмет ранних проявлений; генетическое тестирование более рентабельно, чем ректороманоскопия, для определения того, кто в семье затронут; люди с диагнозом APC - ассоциированные с полипозом состояния в результате наличия у пораженного родственника значительно большей продолжительности жизни, чем у лиц, диагностированных на основании симптомов. Поскольку мониторинг толстой кишки для лиц с риском классического FAP начинается уже в возрасте от 10 до 12 лет, молекулярная генетическое тестирование обычно предлагается детям из группы риска по классическому ФАП в возрасте до десяти лет. Генетическое тестирование при рождении также может быть оправданным, поскольку некоторые родители и педиатры могут рассматривать возможность скрининга гепатобластомы у пораженного потомства с младенчества до пятилетнего возраста. Нет доказательств, указывающих на то, что оптимальный возраст для начала скрининга ". | См. FAP. Также «Скрининг толстой кишки для людей с ослабленным FAP начинается в возрасте от 18 до 20 лет; таким образом, молекулярно-генетическое тестирование должно быть предложено тем, кто подвержен риску аттенуированного FAP примерно в возрасте 18 лет». | ? |
| Наследование и последствия подтвержденного диагноза для других близких родственников | Состояние полипоза, связанного с APC, наследуется по аутосомно-доминантному типу. Примерно 20-25% имеют измененный ген в результате мутации гена de novo. Мало или совсем нет доказательств предвзятости со стороны матери / отца или эффекта, связанного с пожилым отцовским возрастом, в de novo мутации. У братьев и сестер классический 50% риск разделить заболевание, если он унаследован, а не de novo и «низкий», но немного более высокий риск, чем общий, если de novo, поэтому следует предложить генетическое тестирование. У каждого потомства шанс наследования составляет 50%. Другие члены семьи подвергаются риску, если их родители имеют ту же мутацию. Мозаицизм зародышевых линий был зарегистрирован в бессимптомных случаях. Возможно пренатальное тестирование при извлечении плода ДНК. | См. FAP | ? |
Регистры полипоза
Из-за генетической природы FAP реестры полипозов были разработаны во всем мире. Целью этих реестров является повышение осведомленности о передаваемости FAP, а также документирование, отслеживание и уведомление членов семей пострадавших лиц. Одно исследование показало, что использование реестра для уведомления членов семьи (звонков) значительно снижает смертность по сравнению с пробанды.[8] В Регистр полипоза Святого Марка самый старый в мире, начатый в 1924 году, и многие другие регистры полипоза теперь существуют.
Смотрите также
- Колоректальный рак
- Полип (лекарство)
- Аденома
- Аденоматозные полипы
- Колоректальный полип
- Генетическое тестирование
Рекомендации
- ^ а б c d е ж грамм Soravia, C .; Берк, Т .; Мадленский, Л .; Mitri, A .; Cheng, H .; Gallinger, S .; Cohen, Z .; Бапат, Б. (июнь 1998 г.). «Генотип-фенотипическая корреляция в аттенуированной аденоматозной кишечной палочке». Являюсь. J. Hum. Genet. 62 (6): 1290–1301. Дои:10.1086/301883. ЧВК 1377162. PMID 9585611.
- ^ а б c d е ж грамм GeneReviews NBK1345
- ^ а б Семейный аденоматозный полипоз в NLM Домашний справочник по генетике
- ^ Гарднер EJ (июнь 1951 г.). «Генетическое и клиническое исследование полипоза кишечника, предрасполагающего фактора для рака толстой и прямой кишки». Am J Hum Genet. 3 (2): 167–76. ЧВК 1716321. PMID 14902760.
- ^ Амос-Ландграф Дж., Квонг Л.Н., Дав В.Ф. и др. (2007). «Родство крыс с мутантным Apc, выбранным мишенью, усиливает моделирование семейного рака толстой кишки человека». Proc. Natl. Акад. Sci. СОЕДИНЕННЫЕ ШТАТЫ АМЕРИКИ. 104 (10): 4036–41. Дои:10.1073 / pnas.0611690104. ЧВК 1805486. PMID 17360473.
- ^ Джардиелло Ф.М., Круш А.Дж., Петерсен Г.М., Букер С.В., Керр М., Тонг Л.Л., Гамильтон С.Р. (июнь 1994 г.). «Фенотипическая изменчивость семейного аденоматозного полипоза в 11 неродственных семьях с идентичной мутацией гена APC». Гастроэнтерология. 106 (6): 1542–7. Дои:10.1016/0016-5085(94)90408-1. PMID 8194700.
- ^ Hes FJ, Nielsen M, Bik EC, Konvalinka D, Wijnen JT, Bakker E, Vasen HF, Breuning MH, Tops CM (январь 2008 г.). «Соматический мозаицизм APC: недооцененная причина полипоза кишечной палочки». Кишечник. 57 (1): 71–6. Дои:10.1136 / gut.2006.117796. PMID 17604324.
- ^ Рейес Морено Дж., Джинард Висенс Д., Ванрелл М. и др. (2007). «[Влияние реестра на выживаемость семейного аденоматозного полипоза.]». Medicina Clínica (на испанском). 129 (2): 51–2. PMID 17588361.
дальнейшее чтение
- Ясперсон, Кори В .; Patel, Swati G .; Анен, Деннис Дж. (2 февраля 2017 г.). «Состояния полипоза, ассоциированного с APC». В Адаме MP; Ardinger HH; Pagon RA; Уоллес С.Е. (ред.). GeneReviews. Сиэтл, Вашингтон: Вашингтонский университет. PMID 20301519. NBK1345. - полное клиническое резюме FAP и ослабленного FAP, включая пожизненные риски, эпидемиологию и т. Д.
- Семейный аденоматозный полипоз в NLM Домашний справочник по генетике
- Семейный аденоматозный полипоз —eMedicine Гастроэнтерология
- Толстая кишка, синдромы полипоза
- Национальный институт рака: сводная информация о генетике колоректального рака
внешняя ссылка
| Классификация | |
|---|---|
| Внешние ресурсы |