Тромботическая тромбоцитопеническая пурпура - Thrombotic thrombocytopenic purpura
| Тромботическая тромбоцитопеническая пурпура | |
|---|---|
| Другие имена | Синдром Московица,[1] идиопатическая тромботическая тромбоцитопеническая пурпура[2] |
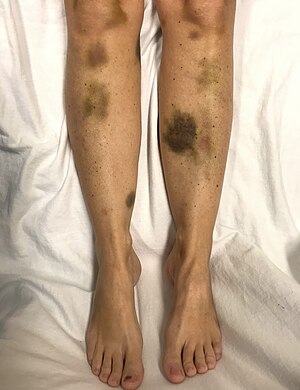 | |
| Самопроизвольный синяк у женщины с критически низким уровнем тромбоцитов | |
| Специальность | Гематология |
| Симптомы | Большие синяки, высокая температура, слабое место, одышка, спутанность сознания, Головная боль[3][2] |
| Обычное начало | Совершеннолетие[3] |
| Причины | Неизвестно, бактериальные инфекции, некоторые лекарства, аутоиммунные заболевания, беременность[3] |
| Диагностический метод | На основании симптомов и анализов крови[2] |
| Дифференциальная диагностика | Гемолитико-уремический синдром (HUS), атипичный гемолитико-уремический синдром (AHUS)[4] |
| Уход | Плазменный обмен, иммунодепрессанты[1] |
| Прогноз | <20% риск смерти[1] |
| Частота | 1 из 100 000 человек[3] |
Тромботическая тромбоцитопеническая пурпура (TTP) это заболевание крови что приводит к сгустки крови формирование в малых кровеносный сосуд по всему телу.[2] Это приводит к низкое количество тромбоцитов, низкие эритроциты из-за их распада, и часто почка, сердце, и мозг дисфункция.[1] Симптомы могут включать: большие синяки, высокая температура, слабое место, одышка, путаница и Головная боль.[2][3] Возможны повторные эпизоды.[3]
Примерно в половине случаев триггер идентифицируется, а в остальных причина остается неизвестной.[3] Известные триггеры включают бактериальные инфекции, некоторые лекарства, аутоиммунные заболевания такие как волчанка, и беременность.[3] Базовый механизм обычно включает антитела подавление фермент ADAMTS13.[1] Это приводит к уменьшению разрушения больших мультимеры из фактор фон Виллебранда (vWF) на более мелкие единицы.[1] Реже ВДП бывает унаследованный от родителей человека, известный как Синдром Апшоу-Шульман, так что дисфункция ADAMTS13 присутствует с рождения.[5] Диагноз обычно основывается на симптомах и анализах крови.[2] Это может быть подтверждено измерением активности или антител против ADAMTS13.[2]
С участием плазмаферез риск смерти снизился с более чем 90% до менее чем 20%.[1] Иммунодепрессанты, такие как глюкокортикоиды, и ритуксимаб также могут быть использованы.[3] Переливание тромбоцитов обычно не рекомендуются.[6]
Пострадало около 1 человека на 100 000 человек.[3] Начало обычно происходит в зрелом возрасте, и чаще страдают женщины.[3] Около 10% случаев начинаются в детстве.[3] Состояние было впервые описано Эли Московиц в 1924 г.[3] Основной механизм был определен в 1980-х и 1990-х годах.[3]
Признаки и симптомы
Признаки и симптомы ТТП сначала могут быть незаметными и неспецифическими. Многие люди испытывают гриппоподобный или диарейное заболевание до развития ТТП.[7] Неврологические симптомы очень распространены и сильно различаются по степени тяжести. Часто сообщаемые симптомы включают: чувствую себя очень уставшим, спутанность сознания, и головные боли.[7] Судороги и симптомы, похожие на симптомы Инсульт также можно увидеть.[7] Другие симптомы включают, помимо прочего, желтуху или бледность кожи, учащенное сердцебиение или одышку, а также пурпурные или красноватые точки на коже небольшого размера, известные как петехии.[нужна цитата ]
По мере прогрессирования ТТП в мелких кровеносных сосудах (микрососуды) образуются тромбы, и тромбоциты (клетки свертывания) расходуются. В результате могут возникнуть синяки, а в редких случаях - кровотечения. Синяк часто принимает форму пурпура, в то время как наиболее частое кровотечение, если оно происходит, - из носа или десен. Более крупные синяки (синяки ) также может развиваться.[нужна цитата ]Классическое проявление ВДП, которое встречается менее чем у 10% людей, включает пять медицинских признаков.[3] Это:
- Высокая температура
- Изменения психического статуса
- Тромбоцитопения
- Снижение функции почек
- Гемолитическая анемия (микроангиопатическая гемолитическая анемия ).[7]
Высокое кровяное давление (гипертония ) можно найти при осмотре.[8]
Причины
ТТП, как и другие микроангиопатические гемолитические анемии (MAHA), вызывается спонтанными агрегирование тромбоцитов и активации коагуляция в мелких кровеносных сосудах. Тромбоциты потребляются в процессе агрегации и связывают vWF. Эти комплексы тромбоцитов и vWF образуют небольшие сгустки крови, которые циркулируют в кровеносных сосудах и вызывают сдвиг эритроцитов, в результате чего их разрыв и формирование шистоциты.[9]Две наиболее понятные причины ТТП связаны с аутоиммунитетом и наследственной недостаточностью ADAMTS13 (известной как синдром Апшоу-Шульмана).[9] Большинство остальных случаев вторично по отношению к какому-то другому фактору.[нужна цитата ]
Аутоиммунный
ВДП неизвестной причины издавна была известна как идиопатический ВДП, но в 1998 году было показано, что большинство случаев вызвано ингибированием фермента. ADAMTS13 к антитела. Отношения редуцированных ADAMTS13 к патогенезу ТТП известна как гипотеза Фурлана-Цая, по названию двух независимых групп исследователей, опубликовавших свои исследования в одном и том же номере журнала. Медицинский журнал Новой Англии.[10][11][12] Эти дела теперь классифицируются как аутоиммунное заболевание и известны как аутоиммунная ТТП (не путать с иммунная / идиопатическая тромбоцитопеническая пурпура ).[нужна цитата ]
ADAMTS13 - это металлопротеиназа несет ответственность за поломку фактор фон Виллебранда (vWF), белок, связывающий тромбоциты, сгустки крови, и стенка кровеносного сосуда в процессе свертывания крови. Очень большие мультимеры vWF более склонны к коагуляции. Следовательно, без надлежащего расщепления vWF с помощью ADAMTS13 коагуляция происходит с более высокой скоростью, особенно в микроциркуляторном русле, части системы кровеносных сосудов, где vWF наиболее активен из-за высокой напряжение сдвига.[5] При идиопатической ТТП сильно сниженная (<5% от нормы) активность ADAMTS13 может быть обнаружена у большинства (80%) людей, и ингибиторы часто встречаются в этой подгруппе (44–56%).
Генетический

Это состояние также может быть врожденным. Такие случаи могут быть вызваны мутациями в гене ADAMTS13.[15] Эта наследственная форма ДТП называется Синдром Апшоу-Шульман.[16][17][18] Люди с наследственным дефицитом ADAMTS13 имеют удивительно мягкий фенотип, но у них развивается ТТП в клинических ситуациях с повышенным уровнем фактора фон Виллебранда, например инфекционное заболевание. Сообщается, что менее 1% всех случаев ТТП вызваны синдромом Апшоу-Шульман.[19] Люди с этим синдромом обычно имеют 5–10% нормальной активности ADAMTS-13.[18][20]
Вторичный
Вторичная ВДП диагностируется, когда в анамнезе человека упоминается одна из известных особенностей, связанных с ВДП. На его долю приходится около 40% всех случаев ТТП. Предрасполагающими факторами являются:[9]
- Рак
- Трансплантация костного мозга
- Беременность
- Медикамент использовать:
- Противовирусные препараты (ацикловир )
- Определенный химиотерапия лекарства, такие как гемцитабин и митомицин С
- Хинин
- Оксиморфон
- Кветиапин
- Бевацизумаб
- Сунитиниб
- Ингибиторы агрегации тромбоцитов (тиклопидин, клопидогрель, и прасугрель )
- Иммунодепрессанты (циклоспорин, митомицин, такролимус / FK506, интерферон-α )
- Гормональные препараты (эстрогены, контрацептивы, заместительная гормональная терапия)[21]
- ВИЧ-1 инфекционное заболевание
Механизм вторичной ТТП плохо изучен, поскольку активность ADAMTS13 обычно не так подавлена, как при идиопатической ТТП, и ингибиторы не могут быть обнаружены. Вероятная этиология может включать, по крайней мере, в некоторых случаях, повреждение эндотелия,[22] хотя образование тромбов, приводящее к окклюзии сосудов, может не иметь существенного значения в патогенезе вторичной ТТП.[23] Эти факторы также можно рассматривать как форму вторичного аГУС; Следовательно, люди с такими особенностями являются потенциальными кандидатами на антикомплементарную терапию.
Механизм
Основной механизм обычно включает опосредованное аутоантителами ингибирование фермент ADAMTS13, а металлопротеиназа отвечает за скалывание больших мультимеров фактор фон Виллебранда (vWF) на более мелкие единицы. Увеличение циркулирующих мультимеров vWF увеличивает адгезию тромбоцитов к участкам эндотелиальный травмы, особенно там, где артериолы и капилляры встречаются, что, в свою очередь, приводит к образованию небольших сгустков тромбоцитов, называемых тромбами. Поскольку тромбоциты используются для образования тромбов, это приводит к снижению общего количества циркулирующих тромбоцитов, что может вызвать опасные для жизни кровотечения. Эритроциты, проходящие через микроскопические сгустки, подвергаются напряжение сдвига, который повреждает их мембраны, что приводит к разрыв красных кровяных телец внутри кровеносных сосудов, что, в свою очередь, приводит к анемия и шистоцит формирование. Наличие этих сгустки крови в мелких кровеносных сосудах уменьшает приток крови к органам, что приводит к повреждению клеток и повреждение конечного органа.[нужна цитата ]
Диагностика
Дифференциальная диагностика
ТТП характеризуется тромботическая микроангиопатия (ТМА) - образование тромбов в мелких кровеносных сосудах по всему телу, что может привести к микроангиопатической гемолитической анемии и тромбоцитопении. Эта характеристика характерна для двух родственных синдромов: гемолитико-уремический синдром (HUS) и атипичный гемолитико-уремический синдром (АГУС).[4] Следовательно, дифференциальная диагностика этих заболеваний, вызывающих ТМА, очень важна. Помимо ТМА, при каждом из этих заболеваний могут присутствовать один или несколько из следующих симптомов: неврологические симптомы (например, спутанность сознания,[24][25] церебральные судороги[25] судороги,[26]); почечная недостаточность[27] (например, повышенный креатинин,[28] снижение расчетной скорости клубочковой фильтрации [рСКФ],[28] аномальный анализ мочи[29]); и желудочно-кишечные (ЖКТ) симптомы (например, диарея[24][30] тошнота / рвота,[26] боль в животе,[26] гастроэнтерит.[24][27] В отличие от HUS и aHUS, TTP, как известно, вызывается приобретенным дефектом в белке ADAMTS13, поэтому лабораторный тест, показывающий ≤5% нормальных уровней ADAMTS13, указывает на TTP.[31] Уровень ADAMTS13 выше 5% в сочетании с положительным тестом на шига-токсин / энтерогеморрагический Кишечная палочка (EHEC), скорее указывают на ГУС,[32] в то время как отсутствие шига-токсина / EHEC может подтвердить диагноз аГУС.[31]
Уход
Из-за высокой смертности от нелеченой ТТП a предполагаемый диагноз ТТП производится даже тогда, когда наблюдаются только микроангиопатическая гемолитическая анемия и тромбоцитопения, и начинается терапия. Переливание крови противопоказано при тромботической ТТП, так как оно способствует развитию коагулопатии. С начала 1990-х гг. плазмаферез стал препаратом выбора при ТТП.[33][34] Это обменное переливание с удалением плазма крови через аферез и замена донорской плазмой (свежезамороженная плазма или криосупернатант ); Процедуру необходимо повторять ежедневно, чтобы устранить ингибитор и уменьшить симптомы. Если аферез недоступен, можно ввести свежезамороженную плазму, но объем, который можно вводить безопасно, ограничен из-за опасности перегрузки жидкостью.[35] Сама по себе инфузия плазмы не так эффективна, как плазмаферез.[33] Кортикостероиды (преднизон или преднизолон ) обычно даются.[34] Ритуксимаб, а моноклональное антитело направленный на CD20 молекула на В-лимфоциты, может использоваться при диагностике; Считается, что это убивает В-клетки и тем самым снижает выработку ингибитора.[34] Более сильная рекомендация для ритуксимаба существует там, где ТТП не отвечает на кортикостероиды и плазмаферез.[34]
Каплацизумаб является альтернативным вариантом лечения ВДП, поскольку было показано, что он способствует более быстрому разрешению болезни по сравнению с людьми, принимавшими плацебо.[36] Однако использование каплацизумаба было связано с увеличением тенденции к кровотечению у исследуемых субъектов.[нужна цитата ]
Люди с рефрактерной или рецидивирующей ТТП могут получать дополнительные иммунодепрессивный терапия, например винкристин, циклофосфамид, циклоспорин А, или же спленэктомия.[3][35]
Дети с синдромом Апшоу-Шульмана получают профилактическую плазму каждые две-три недели; это поддерживает адекватный уровень функционирования ADAMTS13. Некоторые переносят более длительные интервалы между инфузиями плазмы. Дополнительные инфузии плазмы могут потребоваться для запуска событий, таких как хирургическое вмешательство; в качестве альтернативы, количество тромбоцитов можно тщательно контролировать вокруг этих событий, вводя плазму, если количество падает.[37]
Измерения уровня в крови лактатдегидрогеназа, тромбоциты и шистоциты используются для наблюдения за прогрессированием или ремиссией заболевания.[нужна цитата ] Активность ADAMTS13 и уровни ингибитора могут быть измерены во время последующего наблюдения, но у пациентов без симптомов использование ритуксимаба не рекомендуется.[34]
Прогноз
Смертность составляет около 95% для нелеченых случаев, но прогноз достаточно благоприятный (выживание 80–90%) для людей с идиопатической ТТП, у которых диагностирована и лечится рано. плазмаферез.[38]
Эпидемиология
Заболеваемость ТТП составляет около 4–5 случаев на миллион человек в год.[39] Идиопатическая ТТП чаще встречается у женщин и лиц африканского происхождения, а ТТП является вторичной по отношению к аутоиммунным нарушениям, таким как системная красная волчанка чаще встречается у лиц африканского происхождения, хотя другие вторичные формы не показывают этого распределения.[40] Беременные женщины и женщины в послеродовой период составлял заметную часть (12–31%) случаев в некоторых исследованиях; ТТП встречается примерно у одной из 25 000 беременностей.[41]
История
Первоначально ТТП был описан Эли Московиц на Бет Исраэль больница в Нью-Йорк в 1925 г. Московиц приписал болезнь (как теперь известную ошибочно) токсической причине. Московиц отметил, что его пациентка, 16-летняя девочка, страдала анемией, маленький и большие синяки, микроскопические гематурия, а при вскрытии распространен микрососудистые тромбы.[42] В 1966 году обзор 16 новых случаев и 255 ранее зарегистрированных случаев привел к формулированию классической пентады симптомов и результатов (т. Е. Тромбоцитопении, микроангиопатической гемолитической анемии, неврологических симптомов, почечной недостаточности, лихорадки); в этой серии уровень смертности оказался очень высоким (90%).[43]
Хотя реакция на переливание крови отмечалась и раньше, отчет 1978 года и последующие исследования показали, что плазма крови очень эффективна в улучшении процесса заболевания.[44] В 1991 г. сообщалось, что плазмаферез обеспечивает лучшую скорость ответа по сравнению с инфузией плазмы.[45]В 1982 году болезнь была связана с аномально большими мультимерами фактора фон Виллебранда. Выявление недостатка протеаза у людей с ТТП был произведен в 1998-х гг. Местоположение ADAMTS13 в геноме человека было установлено в 2001 году.[44]
Рекомендации
- ^ а б c d е ж грамм Кремер Ховинга, JA; Коппо, П.; Lämmle, B; Moake, JL; Мията, Т; Ванхоорелбеке, К. (6 апреля 2017 г.). «Тромботическая тромбоцитопеническая пурпура». Обзоры природы. Праймеры для болезней. 3: 17020. Дои:10.1038 / nrdp.2017.20. PMID 28382967. S2CID 11960153.
- ^ а б c d е ж грамм «Тромботическая тромбоцитопеническая пурпура приобретенная». Информационный центр по генетическим и редким заболеваниям (GARD) - программа NCATS. Получено 10 октября 2018.
- ^ а б c d е ж грамм час я j k л м п о п Джоли, BS; Коппо, П.; Вейрадье, А (25 мая 2017 г.). «Тромботическая тромбоцитопеническая пурпура». Кровь. 129 (21): 2836–2846. Дои:10.1182 / кровь-2016-10-709857. PMID 28416507.
- ^ а б Джордж Дж. Н. (ноябрь 2010 г.). «Как я лечу пациентов с тромботической тромбоцитопенической пурпурой: 2010». Кровь. 116 (20): 4060–9. Дои:10.1182 / blood-2010-07-271445. PMID 20686117.
- ^ а б Моак JL (2004). «Фактор фон Виллебранда, ADAMTS-13 и тромботическая тромбоцитопеническая пурпура». Семин. Гематол. 41 (1): 4–14. Дои:10.1053 / я.семингематол.2003.10.003. PMID 14727254.
- ^ Вуд, Мари Э .; Филипс, Джордж К. (2003). Секреты гематологии / онкологии. Elsevier Health Sciences. п. 68. ISBN 978-1560535164.
- ^ а б c d Shatzel, JJ; Тейлор, Дж. А. (март 2017 г.). «Синдромы тромботической микроангиопатии». Медицинские клиники Северной Америки (Обзор). 101 (2): 395–415. Дои:10.1016 / j.mcna.2016.09.010. PMID 28189178.
- ^ Allford S, Machin S (2005). «Тромботическая тромбоцитопеническая пурпура». NetDoctor.co.uk.
- ^ а б c Моак Дж. Л. (2002). «Тромботические микроангиопатии». N. Engl. J. Med. 347 (8): 589–600. Дои:10.1056 / NEJMra020528. PMID 12192020.
- ^ Моак JL (1998). «Московиц, мультимеры и металлопротеиназа». N. Engl. J. Med. 339 (22): 1629–31. Дои:10.1056 / NEJM199811263392210. PMID 9828253.
- ^ Фурлан М., Роблес Р., Гальбусера М. и др. (1998). «Протеаза, расщепляющая фактор фон Виллебранда при тромботической тромбоцитопенической пурпуре и гемолитико-уремическом синдроме». N. Engl. J. Med. 339 (22): 1578–84. Дои:10.1056 / NEJM199811263392202. PMID 9828245.
- ^ Цай Х.М., Лян Э.С. (1998). «Антитела к протеазе, расщепляющей фактор фон Виллебранда, при острой тромботической тромбоцитопенической пурпуре». N. Engl. J. Med. 339 (22): 1585–94. Дои:10.1056 / NEJM199811263392203. ЧВК 3159001. PMID 9828246.
- ^ "OMIM Entry - № 274150 - ТРОМБОТИЧЕСКАЯ ТРОМБОЦИТОПЕННАЯ ПУРПУРА, ВРОЖДЕННАЯ; ТТП". www.omim.org. Получено 23 октября 2017.
- ^ ЗАЩИЩЕНО, INSERM US14 - ВСЕ ПРАВА. «Орфанет: тромботическая тромбоцитопеническая пурпура». www.orpha.net. Получено 23 октября 2017.
- ^ Conboy E, Partain PI, Warad D, Kluge ML, Arndt C, Chen D, Rodriguez V (2017) Тяжелый случай врожденной тромботической тромбоцитопении пурпуры, возникшей в результате гетерозиготности соединения с новым патогенным вариантом ADAMTS13. J Педиатр Гематол Онкол
- ^ Шульман И., Пирс М., Люкенс А., Карримбхой З. (июль 1960 г.). «Исследования тромбопоэза. I. Фактор нормальной плазмы человека, необходимый для производства тромбоцитов; хроническая тромбоцитопения из-за его дефицита» (PDF). Кровь. 16 (1): 943–57. Дои:10.1182 / blood.V16.1.943.943. PMID 14443744.
- ^ Апшоу Д.Д. (июнь 1978 г.). «Врожденный дефицит фактора в нормальной плазме, который обращает вспять микроангиопатический гемолиз и тромбоцитопению». N. Engl. J. Med. 298 (24): 1350–2. Дои:10.1056 / NEJM197806152982407. PMID 651994.
- ^ а б Леви Г.Г., Николс В.К., Лиан Е.К., Фоуд Т., Макклинтик Дж. Н., МакГи Б.М., Ян А.Ю., Семениак Д.Р., Старк К.Р., Группа Р., Сароде Р., Шурин С.Б., Чандрасекаран В., Стейблер С.П., Сабио Х., Бухасира Е.Е., Апшоу Д. , Гинзбург Д., Цай Х.М. и др. (Октябрь 2001 г.). «Мутации в члене семейства генов ADAMTS вызывают тромботическую тромбоцитопеническую пурпуру» (PDF). Природа. 413 (6855): 488–494. Bibcode:2001Натура.413..488л. Дои:10.1038/35097008. HDL:2027.42/62592. PMID 11586351. S2CID 4380010.
- ^ Цай Х.М. (2009). «Тромботическая тромбоцитопеническая пурпура, гемолитико-уремический синдром и родственные расстройства». В Greer JP, Foerster J, Rodgers GM et al. (ред.). Клиническая гематология Винтроба (12-е изд.). Филадельфия, Пенсильвания: Липпинкотт, Уильямс и Уилкинс. С. 1314–25. ISBN 978-0781765077.
- ^ Kokame, K .; Мацумото, М; Соедзима, К; Яги, H; Ishizashi, H; Funato, M; Tamai, H; Конно, М; Камиде, К; Кавано, Y; Мията, Т; Фуджимура, Y (14 августа 2002 г.). «Мутации и общие полиморфизмы в гене ADAMTS13, отвечающем за активность протеазы, расщепляющую фактор фон Виллебранда». Proc. Natl. Акад. Sci. Соединенные Штаты Америки. 99 (18): 11902–7. Bibcode:2002PNAS ... 9911902K. Дои:10.1073 / pnas.172277399. ЧВК 129366. PMID 12181489.
- ^ Менкес, Джон Х .; Сарнат, Харви Б .; Мария, Бернард Л. (2006). «Тромботическая тромбоцитопеническая пурпура и гемолитико-уремический синдром». Детская неврология (7-е изд.). Филадельфия: Липпинкотт Уильямс и Уилкинс. п. 525. ISBN 9780781751049.
- ^ van Mourik JA, Boertjes R, Huisveld IA, et al. (Июль 1999 г.). «Пропептид фактора фон Виллебранда при сосудистых заболеваниях: инструмент для различения острых и хронических нарушений эндотелиальных клеток». Кровь. 94 (1): 179–85. Дои:10.1182 / blood.V94.1.179.413k18_179_185. PMID 10381511.
- ^ Ивата Х, Ками М, Хори А., Хамаки Т, Такеучи К., Муто И (июнь 2001 г.). «Ретроспективное исследование вторичной тромботической тромбоцитопенической пурпуры на основе аутопсии». Haematologica. 86 (6): 669–70. PMID 11418383.
- ^ а б c Норис М., Каприоли Дж., Бресин Э. и др. (Октябрь 2010 г.). «Относительная роль генетических аномалий комплемента в спорадических и семейных аГУС и их влияние на клинический фенотип». Clin J Am Soc Nephrol. 5 (10): 1844–59. Дои:10.2215 / CJN.02210310. ЧВК 2974386. PMID 20595690.
- ^ а б Neuhaus TJ, Calonder S, Leumann EP (июнь 1997 г.). «Гетерогенность атипичных гемолитико-уремических синдромов». Arch. Дис. Ребенок. 76 (6): 518–21. Дои:10.1136 / adc.76.6.518. ЧВК 1717216. PMID 9245850.
- ^ а б c Дракон-Дурей М.А., Сетхи С.К., Багга А. и др. (Декабрь 2010 г.). «Клинические особенности гемолитико-уремического синдрома, ассоциированного с аутоантителами против фактора Н». Варенье. Soc. Нефрол. 21 (12): 2180–7. Дои:10.1681 / ASN.2010030315. ЧВК 3014031. PMID 21051740.
- ^ а б Каприоли Дж., Норис М., Бриоши С. и др. (Август 2006 г.). «Генетика HUS: влияние мутаций MCP, CFH и IF на клинические проявления, ответ на лечение и исход». Кровь. 108 (4): 1267–79. Дои:10.1182 / кровь-2005-10-007252. ЧВК 1895874. PMID 16621965.
- ^ а б Арисета Г., Бесбас Н., Джонсон С. и др. (Апрель 2009 г.). «Руководство по исследованию и начальной терапии диарея-отрицательного гемолитико-уремического синдрома». Педиатр. Нефрол. 24 (4): 687–96. Дои:10.1007 / s00467-008-0964-1. PMID 18800230.
- ^ Аль-Акаш С.И., Алмонд П.С., Савелл В.Х., Гарайбех С.И., Хог С. (апрель 2011 г.). «Экулизумаб вызывает длительную ремиссию при рецидивирующем посттрансплантационном ГУС, связанном с мутацией гена С3». Педиатр. Нефрол. 26 (4): 613–9. Дои:10.1007 / s00467-010-1708-6. PMID 21125405. S2CID 22334044.
- ^ Zuber J, Le Quintrec M, Sberro-Soussan R, Loirat C, Frémeaux-Bacchi V, Legendre C (январь 2011 г.). «Новые взгляды на гемолитико-уремический синдром после трансплантации почки». Нат Рев Нефрол. 7 (1): 23–35. Дои:10.1038 / nrneph.2010.155. PMID 21102542. S2CID 2054556.
- ^ а б Цай Х.М. (январь 2010 г.). «Патофизиология тромботической тромбоцитопенической пурпуры». Int. J. Hematol. 91 (1): 1–19. Дои:10.1007 / s12185-009-0476-1. ЧВК 3159000. PMID 20058209.
- ^ Битзан М., Шефер Ф., Реймонд Д. (сентябрь 2010 г.). «Лечение типичного (энтеропатического) гемолитико-уремического синдрома». Семин. Тромб. Хемост. 36 (6): 594–610. Дои:10.1055 / с-0030-1262881. PMID 20865636.
- ^ а б Майкл, М; Elliott, EJ; Ридли, Г. Ф.; Ходсон, EM; Крейг, JC (21 января 2009 г.). «Вмешательства при гемолитико-уремическом синдроме и тромботической тромбоцитопенической пурпуре». Кокрановская база данных систематических обзоров (1): CD003595. Дои:10.1002 / 14651858.CD003595.pub2. HDL:10072/61440. ЧВК 7154575. PMID 19160220.
- ^ а б c d е Лим В., Веселый СК, Джордж Дж. Н. (5 марта 2015 г.). «Роль ритуксимаба в лечении пациентов с приобретенной тромботической тромбоцитопенической пурпурой». Кровь. 125 (10): 1526–31. Дои:10.1182 / кровь-2014-10-559211. ЧВК 4351502. PMID 25573992.
- ^ а б Allford SL, Hunt BJ, Rose P, Machin SJ (февраль 2003 г.). «Рекомендации по диагностике и лечению тромботических микроангиопатических гемолитических анемий». Br. J. Haematol. 120 (4): 556–73. Дои:10.1046 / j.1365-2141.2003.04049.x. PMID 12588343. S2CID 38774829.
- ^ Пейванди, Флора; Скалли, Мари; Кремер Ховинга, Йоханна А .; Каталанд, Сперо; Knöbl, Пауль; Ву, Хайфэн; Артони, Андреа; Вествуд, Джон-Пол; Мансури Талегани, Магнус (11 февраля 2016 г.). «Каплацизумаб при приобретенной тромботической тромбоцитопенической пурпуре» (PDF). Медицинский журнал Новой Англии. 374 (6): 511–522. Дои:10.1056 / NEJMoa1505533. ISSN 0028-4793. PMID 26863353.
- ^ Loirat, C; Girma, JP; Desconclois, C; Коппо, П.; Вейрадье, А (январь 2009 г.). «Тромботическая тромбоцитопеническая пурпура, связанная с тяжелым дефицитом ADAMTS13 у детей». Детская нефрология (Берлин, Германия). 24 (1): 19–29. Дои:10.1007 / s00467-008-0863-5. PMID 18574602. S2CID 22209831.
- ^ Цай, Хан-Моу (февраль 2006 г.). «Современные концепции тромботической тромбоцитопенической пурпуры». Ежегодный обзор медицины. 57: 419–436. Дои:10.1146 / annurev.med.57.061804.084505. ЧВК 2426955. PMID 16409158.
- ^ Террелл Д.Р., Уильямс Л.А., Веселы С.К., Ляммле Б., Ховинга Дж. А., Джордж Дж. Н. (июль 2005 г.). «Частота тромботической тромбоцитопенической пурпуры-гемолитико-уремического синдрома: все пациенты, идиопатические пациенты и пациенты с тяжелым дефицитом ADAMTS-13». J. Thromb. Haemost. 3 (7): 1432–6. Дои:10.1111 / j.1538-7836.2005.01436.x. PMID 15978100.
- ^ Террелл Д.Р., Веселы С.К., Кремер Ховинга Я.А., Ляммле Б., Джордж Дж. Н. (ноябрь 2010 г.). «Различные различия пола и расы среди тромботической тромбоцитопенической пурпуры и гемолитико-уремических синдромов». Являюсь. J. Hematol. 85 (11): 844–7. Дои:10.1002 / ajh.21833. ЧВК 3420337. PMID 20799358.
- ^ X. Лун Чжэн; Дж. Эван Сэдлер (2008). «Патогенез тромботических микроангиопатий». Ежегодный обзор патологии. 3: 249–277. Дои:10.1146 / annurev.pathmechdis.3.121806.154311. ЧВК 2582586. PMID 18215115.
- ^ Moschcowitz E (1924). «Острая фебрильная плейохромная анемия с гиалиновым тромбозом терминальных артериол и капилляров: неописанное заболевание». Proc NY Pathol Soc. 24: 21–4. Перепечатано в Moschcowitz E (октябрь 2003 г.). «Острая фебрильная плейохромная анемия с гиалиновым тромбозом терминальных артериол и капилляров: неописанное заболевание. 1925». Mt. Синай Дж. Мед. 70 (5): 352–5. PMID 14631522..
- ^ Амороси Э.Л., Ультманн Дж.Э. (1966). «Тромбоцитопическая пурпура: отчет о 16 случаях и обзор литературы». Медицина (Балтимор). 45 (2): 139–159. Дои:10.1097/00005792-196603000-00003. S2CID 71943329.
- ^ а б Сэдлер, Дж. Э. (2008). «Фактор фон Виллербранда, ADAMTS13 и тромботическая тромбоцитопеническая пурпура». Кровь. 112 (1): 11–18. Дои:10.1182 / кровь-2008-02-078170. ЧВК 2435681. PMID 18574040.
- ^ Рок Г.А., Шумак К.Х., Бускард Н.А. и др. (Август 1991 г.). «Сравнение плазмафереза с инфузией плазмы при лечении тромботической тромбоцитопенической пурпуры. Канадская группа изучения афереза». N. Engl. J. Med. 325 (6): 393–7. Дои:10.1056 / NEJM199108083250604. PMID 2062330.
внешняя ссылка
| Классификация | |
|---|---|
| Внешние ресурсы |