Сульфоновая кислота - Sulfonic acid

А сульфоновая кислота (или сульфоновая кислота) относится к члену класса сероорганические соединения с общей формулой R − S (= O)2−OH, где R - органический алкил или арил группа и S (= O)2(ОН) группа а сульфонил гидроксид.[1] В качестве заместителя он известен как сульфогруппа. Сульфоновую кислоту можно рассматривать как серная кислота с одной гидроксильной группой, замещенной органической заместитель. В исходное соединение (с органическим заместителем, замещенным водородом) - исходная сульфоновая кислота, HS (= O)2(ОН), а таутомер из сернистая кислота, S (= O) (ОН)2.[2] Соли или сложные эфиры сульфоновых кислот называются сульфонаты.
Подготовка

Сульфоновые кислоты производятся путем сульфирование. Обычно сульфирующим агентом является триоксид серы. Широкомасштабным применением этого метода является производство алкилбензолсульфоновые кислоты:
- RC6ЧАС5 + ТАК3 → RC6ЧАС4ТАК3ЧАС
В этой реакции триоксид серы является электрофил и арена подвергается электрофильное ароматическое замещение.[1] Прямое сульфирование также преобразует метансульфоновая кислота к метандисульфоновая кислота.
Многие алкансульфоновые кислоты получают из бисульфит, который присоединяется к терминальным алкенам или алкилированный алкилгалогенидами:[3]
- HSO3− + RCH = CH2 + H+ → RCH2CH2ТАК3ЧАС
- HSO3− + RBr → RSO3H + Br−
Сульфоновые кислоты можно получить окислением тиолов:
- RSH +3⁄2 О2 → RSO3ЧАС
Такой путь лежит в основе биосинтеза таурин.
Маршруты гидролиза
Многие сульфоновые кислоты получают гидролизом сульфонилгалогенидов и родственных предшественников. Таким образом, перфтороктансульфоновая кислота получают путем гидролиза сульфонилфторида, который, в свою очередь, образуется электрофторирование октансульфоновой кислоты. Аналогичным образом сульфонилхлорид, полученный из полиэтилена, гидролизуется до сульфоновой кислоты. Эти сульфонилхлориды образуются в результате свободнорадикальных реакций хлора, диоксида серы и углеводородов с использованием Рид реакция.
Винилсульфоновая кислота получается путем гидролиза карбилсульфат, (C2ЧАС4(ТАК3)2), который, в свою очередь, получают добавлением триоксида серы к этилен.
Свойства
Сульфоновые кислоты - сильные кислоты. Их обычно называют примерно в миллион раз сильнее, чем соответствующие карбоновый кислота. Например, п-Толуолсульфоновая кислота и метансульфоновая кислота имеют пKа значения −2,8 и −1,9 соответственно, а бензойная кислота и уксусная кислота равны 4,20 и 4,76 соответственно. Однако из-за их высокой кислотности их pKа значения не могут быть измерены напрямую, и обычно приводимые значения следует рассматривать как косвенные оценки со значительной неопределенностью. Например, в различных источниках сообщается, что рKа метансульфоновой кислоты до -0,6[4] или всего -6,5.[5] Известно, что сульфоновые кислоты реагируют с твердым хлоридом натрия (поваренная соль ) с образованием сульфоната натрия и хлористого водорода.[6] Это свойство подразумевает кислотность в пределах двух или трех порядков от кислотности HCl.(г), чей pKа был недавно точно определен (pKаводный = −5.9).
Из-за своей полярности сульфоновые кислоты обычно представляют собой твердые кристаллические вещества или вязкие высококипящие жидкости. Они также обычно бесцветны и не окисляют.[7] что делает их пригодными для использования в качестве кислотных катализаторов в органических реакциях. Их полярность в сочетании с их высокой кислотностью делает короткоцепочечные сульфоновые кислоты водорастворимыми, в то время как длинноцепочечные сульфоновые кислоты проявляют свойства, подобные детергентам.
Строение сульфоновых кислот поясняется прототипом, метансульфоновая кислота. Группа сульфоновой кислоты, RSO2ОН имеет тетраэдрический серный центр, что означает, что сера находится в центре четырех атомов: трех атомов кислорода и одного углерода. Общая геометрия серного центра напоминает форму серная кислота.
- Типичные сульфоновые кислоты и сульфонаты
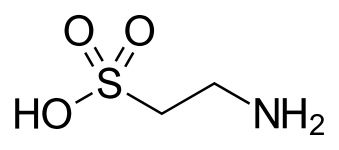
Таурин, а желчная кислота, и одна из немногих природных сульфоновых кислот (показана в необычных таутомер ).

ПФОС, поверхностно-активное вещество и загрязнитель, вызывающий споры.

п-Толуолсульфоновая кислота, широко используемый реагент в органическом синтезе.

Нафион, полимерная сульфоновая кислота, полезная в топливные элементы.
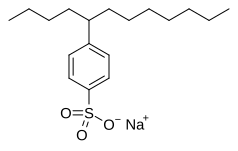
Додецилбензолсульфонат натрия, алкилбензолсульфонат поверхностно-активное вещество используется в стиральные порошки.

Коэнзим-М, является кофактором, необходимым для биосинтеза метан, нашел в натуральный газ.






Приложения
Хотя известны как алкил, так и арилсульфоновые кислоты, большинство их применений связано с ароматическими производными.
Моющие и поверхностно-активные вещества
Моющие средства и поверхностно-активные вещества представляют собой молекулы, которые объединяют сильно неполярные и высокополярные группы. Традиционно мыло популярные поверхностно-активные вещества, полученные из жирные кислоты. С середины 20 века использование сульфоновых кислот превзошло использование мыла в развитых обществах. Например, примерно 2 миллиарда килограммов алкилбензолсульфонаты выпускаются ежегодно для различных целей. Сульфонаты лигнина, полученные сульфированием лигнин входят в состав буровых растворов и добавок в определенные виды бетон.[8]
Красители
Многие, если не большинство антрохинон красители производятся или обрабатываются сульфированием.[9] Сульфоновые кислоты имеют тенденцию плотно связываться с белки и углеводы. Самый «моющийся» красители являются сульфоновыми кислотами (или имеют функциональные сульфонил группа в них) по этой причине. п-Крезидинсульфоновая кислота используется для изготовления пищевых красителей.
Кислотные катализаторы
Сульфоновые кислоты, являясь сильными кислотами, также используются в качестве катализаторы. Самые простые примеры: метансульфоновая кислота, CH3ТАК2ОН и п-толуолсульфоновая кислота, которые регулярно используются в органическая химия как кислоты, которые являются липофильными (растворимы в органических растворителях). Также можно использовать полимерные сульфоновые кислоты. Dowex смолы являются производными сульфоновой кислоты полистирол и используется как катализаторы и для ионного обмена (умягчение воды ). Нафион, фторированная полимерная сульфоновая кислота является компонентом протонообменных мембран в топливные элементы.[10]
Наркотики
Сульфаниламидные препараты, класс антибактериальных средств, производятся из сульфоновых кислот.

Лигносульфонаты
в сульфитный процесс для изготовления бумаги лигнин удаляют из лигноцеллюлозы путем обработки древесной стружки растворами сульфитных и бисульфит-ионов. Эти реагенты расщепляют связи между компонентами целлюлозы и лигнина, особенно внутри самого лигнина. Лигнин превращается в лигносульфонаты, полезно иономеры, которые растворимы и могут быть отделены от целлюлозных волокон.

Реакции
Гидролиз
Арилсульфоновые кислоты подвержены гидролизу, обратному реакции сульфирования. В то время как бензолсульфоновая кислота гидролизуется при температуре выше 200 ° C, большинство родственных производных гидролизуются легче. Таким образом, нагревание арилсульфоновых кислот в водной кислоте дает исходный арен. Эта реакция используется в нескольких сценариях. В некоторых случаях сульфоновая кислота служит в качестве водорастворимой защитной группы, как показано на примере очистки пара-ксилола через его производное сульфоновой кислоты. Синтез 2,6-дихлорфенол, фенол превращается в его производное 4-сульфоновой кислоты, которое затем селективно хлорируется в положениях, фланкирующих фенол. Гидролиз высвобождает группу сульфоновой кислоты.[11]
Этерификация
Сульфоновые кислоты могут быть преобразованы в сложные эфиры. Этот класс органические соединения имеет общую формулу R − SO2-ИЛИ. Сульфоновые эфиры, такие как метилтрифлат считаются хорошими алкилирующие агенты в органический синтез. Такие сложные эфиры сульфоновой кислоты часто получают алкоголиз сульфонилхлоридов:
- RSO2Cl + R′OH → RSO2OR ′ + HCl
Галогенирование
Сульфонилгалогенидные группы возникают, когда сульфонильная функциональная группа однократно связана с атомом галогена. У них есть общая формула R − SO2−X, где X - галогенид, почти всегда хлорид. Их получают хлорированием сульфоновых кислот с использованием тионилхлорид и соответствующие реагенты.
Вытеснение гидроксидом
Хотя (арил) C − SO3− связь может быть разорвана нуклеофильными реагентами. Историческое и непреходящее значение имеет α-сульфирование антрохинона с последующим замещением сульфонатной группы другими нуклеофилами, что не может быть установлено напрямую.[9] Ранний метод производства фенол участвовал в основном гидролизе натрия бензолсульфонат, который легко образуется из бензола.[12]
- C6ЧАС5ТАК3Na + NaOH → С6ЧАС5ОН + Na2ТАК3
Однако условия для этой реакции суровые, требующие наличия «плавленой щелочи» или расплавленного гидроксида натрия при 350 ° C для самой бензолсульфоновой кислоты.[13] В отличие от механизма конденсированного щелочного гидролиза хлорбензола, который протекает через элиминирование-присоединение (бензин механизм), бензолсульфоновая кислота претерпевает аналогичное превращение под действием SNМеханизм Ar, как показал 14С-мечение, несмотря на отсутствие стабилизирующих заместителей.[14] Сульфоновые кислоты с электроноакцепторными группами (например, с NO2 или заместители CN) гораздо легче претерпевают это превращение.
использованная литература
- ^ а б Марш, Джерри (1992), Продвинутая органическая химия: реакции, механизмы и структура (4-е изд.), Нью-Йорк: Wiley, ISBN 0-471-60180-2
- ^ Ни исходная сульфоновая кислота, ни исходная сернистая кислота не были выделены и даже не наблюдались, хотя моноанион этих гипотетических частиц существует в растворе в виде равновесной смеси таутомеров: HS (= O)2(O⊖) ⇌S (= O) (OH) (O⊖).
- ^ Коссвиг, Курт (2000). «Сульфоновые кислоты алифатические». Энциклопедия промышленной химии Ульмана. Вайнхайм: Wiley-VCH. Дои:10.1002 / 14356007.a25_503.
- ^ Бордвелл, Фредерик Г. (1988). «Равновесные кислотности в растворе диметилсульфоксида». Отчеты о химических исследованиях. 21 (12): 456–463. Дои:10.1021 / ar00156a004. ISSN 0001-4842.
- ^ Смит, Майкл; Март, Джерри (2007). Продвинутая органическая химия марта: реакции, механизмы и структура (6-е изд.). Хобокен, штат Нью-Джерси: Wiley-Interscience. ISBN 9781615838424. OCLC 708034394.
- ^ Клейден, Джонатан; Гривс, Ник; Уоррен, Стюарт Г. Органическая химия (2-е изд.). Оксфорд. ISBN 9780191666216. OCLC 867050415.
- ^ Гернон, Майкл Д .; Ву, Мин; Бушта, Томас; Дженни, Патрик (1999). «Экологические преимущества метансульфоновой кислоты». Зеленая химия. 1 (3): 127–140. Дои:10.1039 / A900157C. ISSN 1463-9262.
- ^ Косвиг, К. «Поверхностно-активные вещества» в Энциклопедии промышленной химии Ульмана, 2002 г., Wiley-VCH, Weinheim. Дои:10.1002 / 14356007.a25_747.
- ^ а б Бьен, Ханс-Самуэль; Ставиц, Йозеф; Вундерлих, Клаус (2002). «Антрахиноновые красители и промежуточные продукты». Энциклопедия промышленной химии Ульмана. Вайнхайм: Wiley-VCH. Дои:10.1002 / 14356007.a02_355.
- ^ Буска, Гвидо (2007). «Кислотные катализаторы в промышленной химии углеводородов». Chem. Rev. 107: 5366–5410. Дои:10.1021 / cr068042e.
- ^ Отто Линднер, Ларс Родефельд (2005). «Бензолсульфоновые кислоты и их производные». Энциклопедия промышленной химии Ульмана. Вайнхайм: Wiley-VCH. Дои:10.1002 / 14356007.a03_507.CS1 maint: использует параметр авторов (ссылка на сайт)
- ^ Манфред Вебер, Маркус Вебер, Майкл Кляйне-Бойманн «Фенол» в Энциклопедии промышленной химии Ульмана 2004, Wiley-VCH. Дои:10.1002 / 14356007.a19_299.pub2.
- ^ Баннетт, Джозеф Ф .; Захлер, Роланд Э. (1951-10-01). «Реакции ароматического нуклеофильного замещения». Химические обзоры. 49 (2): 273–412. Дои:10.1021 / cr60153a002. ISSN 0009-2665.
- ^ Оэ, Сигеру; Фурукава, Наомити; Кисе, Масахиро; Каваниси, Мицуёси (1966). «Механизм щелочного плавления бензолсульфоновой кислоты». Бюллетень химического общества Японии. 39 (6): 1212–1216. Дои:10.1246 / bcsj.39.1212.
| Авторитетный контроль |
|---|