Синдром удлиненного интервала QT - Long QT syndrome
| Синдром удлиненного интервала QT | |
|---|---|
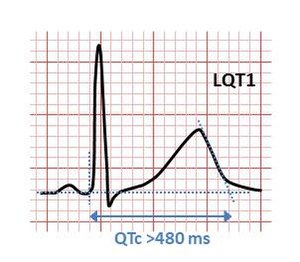 | |
| ЭКГ показывает типичную картину наследственного синдрома удлиненного интервала QT (LQT1). Интервал QT> 480 мс считается ненормально длинным. | |
| Специальность | Кардиология |
| Симптомы | Обморок, потеря слуха, припадки[1] |
| Осложнения | Внезапная смерть[1] |
| Причины | Генетический, некоторые лекарства, низкий уровень калия в крови, низкий уровень кальция в крови, сердечная недостаточность[2] |
| Факторы риска | Семейный анамнез внезапной смерти[3] |
| Диагностический метод | ЭКГ (ЭКГ), клинические данные, генетическое тестирование [4][5] |
| Дифференциальный диагноз | Синдром Бругада, аритмогенная дисплазия правого желудочка[3] |
| лечение | Избегать физических упражнений, получать достаточно калий, бета-блокаторы, имплантируемый кардиодефибриллятор[6] |
| Частота | ≈ 1 из 7000[6] |
| Летальные исходы | ≈3 500 в год (США)[6] |
Синдром удлиненного интервала QT (LQTS) - состояние, при котором реполяризация из сердце после стук сердца подвергается воздействию.[7] Это приводит к повышенному риску аритмия что может привести к обморок, тонущий, припадки, или внезапная смерть.[1] Эти эпизоды могут быть вызваны физическими упражнениями или стрессом.[6] Некоторые редкие формы LQTS связаны с другими симптомами и признаками, включая: глухота и периоды мышечной слабости. [1]
Синдром удлиненного интервала QT может присутствовать при рождении или развиваться в более позднем возрасте.[1] В унаследованный форма может возникать сама по себе или как часть более крупного генетическое расстройство.[1] Возникновение в более позднем возрасте может быть результатом приема определенных лекарств, низкий уровень калия в крови, низкий уровень кальция в крови, или сердечная недостаточность.[2] Лекарства, к которым причастны, включают определенные антиаритмические средства, антибиотики, и нейролептики.[2] LQTS можно диагностировать с помощью ЭКГ (ЭКГ), если скорректированный интервал QT более 480–500 миллисекунд, но клинические данные, другие характеристики ЭКГ и генетическое тестирование может подтвердить диагноз с более короткими интервалами QT.[4][5]
Управление может включать в себя отказ от физических упражнений, получение достаточного калий в диете употребление бета-блокаторы, или имплантируемый кардиодефибриллятор.[6] Для людей с LQTS, которые выживают остановка сердца и остаются без лечения, риск смерти в течение 15 лет превышает 50%.[8][6] При правильном лечении этот показатель снижается до менее 1% за 20 лет.[3]
Синдром удлиненного интервала QT встречается у 1 из 7000 человек.[6] Женщины страдают чаще, чем мужчины.[6] У большинства людей симптомы заболевания развиваются до достижения ими 40-летнего возраста.[6] Это относительно частая причина внезапной смерти наряду с Синдром Бругада и аритмогенная дисплазия правого желудочка.[3] В Соединенных Штатах от него умирает около 3500 человек в год.[6] Впервые это состояние было четко описано в 1957 году.[9]
Признаки и симптомы
Многие люди с синдромом удлиненного интервала QT не имеют никаких признаков или симптомов. Когда возникают симптомы, они обычно вызваны: аномальные сердечные ритмы (аритмии), чаще всего форма вентрикулярная тахикардия называется Торсады-де-пуанты (TdP). Если аритмия переходит в нормальный ритм спонтанно у пострадавшего может возникнуть головокружение (известное как пресинкоп ) или слабый которому может предшествовать ощущение трепетания в груди.[6] Если аритмия продолжается, у пострадавшего может наблюдаться остановка сердца, которые при отсутствии лечения могут привести к внезапной смерти.[10] Те, у кого LQTS, могут также испытать судорожная активность (неэпилептический припадок) в результате снижения притока крови к мозгу во время аритмии.[11][12] Эпилепсия также связан с некоторыми типами синдрома удлиненного интервала QT.[12]
Аритмии, приводящие к обморокам и внезапной смерти, чаще возникают при определенных обстоятельствах, частично определяемых тем, что генетический вариант настоящее. Хотя аритмии могут возникать в любое время, при некоторых формах LQTS аритмии чаще наблюдаются в ответ на физическую нагрузку или психическое напряжение (LQT1), в других формах после внезапного громкого шума (LQT2) и в некоторых формах во время сна или сразу после бодрствование (LQT3).[10][13]
Некоторые редкие формы синдрома удлиненного интервала QT поражают другие части тела, что приводит к глухота в Джервелл и Ланге-Нильсен форма условия, и периодический паралич в Андерсен-Тавил (LQT7) форма.[4]
Риск аритмий
В то время как у людей с синдромом удлиненного интервала QT повышен риск развития нарушений сердечного ритма, абсолютный риск аритмий очень варьируется.[14] Самый надежный предиктор того, разовьется ли у кого-то TdP, - это пережили ли они в прошлом эту аритмию или другую форму остановки сердца.[15] Те с LQTS, которые испытали обморок без записи ЭКГ, также подвержены более высокому риску, поскольку обмороки в этих случаях часто возникают из-за недокументированной самоустанавливающейся аритмии.[15]
Помимо аритмий в анамнезе, степень удлинения интервала QT позволяет прогнозировать риск. В то время как у некоторых интервалы QT очень удлинены, у других наблюдается лишь небольшое удлинение QT или даже нормальный интервал QT в покое (скрытый LQTS). Те, у кого самые длинные интервалы QT, с большей вероятностью будут испытывать TdP, а скорректированный интервал QT более 500 мс, как полагают, представляет людей с более высоким риском.[16] Несмотря на это, пациенты с незначительным удлинением интервала QT или скрытым LQTS все же имеют некоторый риск аритмий.[10] В целом, каждые 10 мс увеличение скорректированного интервала QT связано с увеличением риска аритмии на 5%.[14]
Поскольку эффекты удлинения QT как генетических вариантов, так и приобретенных причин LQTS являются аддитивными, люди с наследственным LQTS с большей вероятностью будут испытывать TdP, если им назначены препараты, удлиняющие QT, или если они испытывают проблемы с электролитом например, низкий уровень калия в крови (гипокалиемия ). Точно так же те, кто принимает препараты, удлиняющие QT, с большей вероятностью будут испытывать TdP, если у них есть генетическая тенденция к удлинению интервала QT, даже если эта тенденция скрыта.[14] Аритмии чаще возникают в лекарственно-индуцированный LQTS если лекарство было дано быстро внутривенно, или если в крови человека присутствуют высокие концентрации препарата.[16] Риск аритмий также выше, если у человека, принимающего препарат, сердечная недостаточность, принимает дигиталис, или недавно перенесла кардиоверсию мерцательная аритмия.[16] К другим факторам риска развития torsades de pointes среди лиц с LQTS относятся женский пол, возраст, уже существующие сердечно-сосудистые заболевания, и ненормальный печень или функция почек.[17]
Причины
Существует несколько подтипов синдрома удлиненного интервала QT. Их можно в общих чертах разделить на те, которые вызваны: генетические мутации те, с которыми больные рождаются, переносятся на протяжении всей своей жизни и могут передаваться своим детям (наследственный или врожденный синдром удлиненного интервала QT), а также те, которые вызваны другими факторами, которые не могут передаваться и часто являются обратимыми (синдром приобретенного удлиненного интервала QT).
Унаследовано

Унаследованный, или врожденный синдром удлиненного интервала QT, вызван генетическими аномалиями. LQTS может возникать из вариантов в нескольких генах, что в некоторых случаях приводит к совершенно разным характеристикам.[18] Эти варианты объединяет то, что они влияют на один или несколько ионные токи ведущий к продлению потенциал желудочкового действия, таким образом удлиняя интервал QT.[7] Были предложены системы классификации для различения подтипов состояния на основе клинических признаков (и названных в честь тех, кто впервые описал состояние) и подразделенных по лежащему в основе генетическому варианту.[19] Самым распространенным из них, составляющим 99% случаев, является Синдром Романо-Уорда (генетически LQT1-6 и LQT9-16), аутосомно-доминантная форма, при которой электрическая активность сердца нарушается без вовлечения других органов.[10] Менее распространенной формой является синдром Джервелла и Ланге-Нильсена, аутосомно-рецессивная форма LQTS, сочетающая удлиненный интервал QT с врожденной глухотой.[20] Другие редкие формы включают синдром Андерсона-Тавила (LQT7) с такими особенностями, как удлиненный интервал QT, периодический паралич и аномалии лица и скелета; и Синдром Тимоти (LQT8), при котором удлинение интервала QT связано с аномалиями структуры сердца и расстройство аутистического спектра.[4]
Синдром Романо-Уорда
LQT1 - наиболее распространенный подтип синдрома Романо – Уорда, на который приходится от 30 до 35% всех случаев.[21] Ответственный ген, KCNQ1, был изолирован хромосома 11p 15.5 и кодирует альфа-субъединицу KvLQT1 калиевый канал. Эта субъединица взаимодействует с другими белками (в частности, с субъединицей minK beta), чтобы создать канал, по которому протекает задержанный ток калиевого выпрямителя. яKs отвечает за фазу реполяризации потенциал сердечного действия.[21] Варианты в KCNQ1 это уменьшение яKs (варианты потери функции) замедляют реполяризацию потенциала действия. Это вызывает подтип LQT1 синдрома Романо-Уорда, когда наследуется единственная копия варианта (гетерозиготное, аутосомно-доминантное наследование). Наследование двух копий варианта (гомозиготное, аутосомно-рецессивное наследование) приводит к более тяжелому синдрому Джервелла и Ланге – Нильсена.[21] И наоборот, варианты в KCNQ1, которые увеличивают яKs привести к более быстрой реполяризации и синдром короткого интервала QT.[22]
Подтип LQT2 - вторая по распространенности форма синдрома Романо – Уорда, на которую приходится от 25 до 30% всех случаев.[21] Это вызвано вариантами в KCNH2 ген (также известный как hERG) на хромосоме 7, которая кодирует калиевый канал, по которому проходит быстрый внутренний выпрямительный ток. яKr.[21] Этот ток вносит вклад в конечную фазу реполяризации сердечного потенциала действия и, следовательно, в длину интервала QT.[21]
Подтип LQT3 синдрома Романо-Уорда вызван вариантами в SCN5A ген, расположенный на хромосоме 3p21–24. SCN5A кодирует альфа-субъединицу сердечного натриевого канала, NaV1.5, отвечает за натриевый ток яNa который деполяризует сердечные клетки в начале потенциала действия.[21] Сердечные натриевые каналы обычно быстро инактивируются, но мутации, вовлеченные в LQT3, замедляют их инактивацию, что приводит к небольшому устойчивому «позднему» натриевому току. Этот продолжающийся внутренний ток продлевает потенциал действия и, следовательно, интервал QT.[21] Хотя некоторые варианты в SCN5A вызывают LQT3, другие варианты могут вызывать совершенно другие условия. Варианты, вызывающие снижение раннего пикового тока, могут вызвать Синдром Бругада и заболевание сердечной проводимости, в то время как другие варианты были связаны с дилатационная кардиомиопатия. Некоторые варианты, которые влияют как на ранний, так и на поздний натриевый ток, могут вызывать синдромы перекрытия которые сочетают в себе аспекты как LQT3, так и синдрома Бругада.[10]
Редкие подтипы Романо – Варда (LQT4-6 и LQT9-16)
LQT5 вызван вариантами в KCNE1 ген, ответственный за бета-субъединицу калиевого канала MinK. Эта субъединица в сочетании с альфа-субъединицей, кодируемой KCNQ1, отвечает за калиевый ток. яKs который уменьшается в LQTS.[21] LQT6 вызван вариантами в KCNE2 ген, ответственный за бета-субъединицу MiRP1 калиевого канала, который генерирует калиевый ток яKr.[21] Варианты, которые уменьшают этот ток, связаны с удлинением интервала QT.[20] Однако последующие данные, такие как относительно частое обнаружение вариантов в гене у людей без синдрома удлиненного интервала QT, и общая необходимость наличия второго фактора стресса, такого как гипокалиемия, для выявления удлинения интервала QT, позволили предположить, что этот ген вместо этого представляет собой модификатор предрасположенности к удлинению интервала QT.[19] Поэтому некоторые спорят, есть ли варианты в KCNE2 достаточно, чтобы вызвать синдром Романо-Варда сами по себе.[19]
LQT9 вызывается вариантами структурного белка мембраны, кавеолин -3.[21] Кавеолины образуют определенные мембранные домены, называемые кавеолы в котором находятся потенциалозависимые натриевые каналы. Подобно LQT3, эти варианты кавеолина увеличивают поздний устойчивый натриевый ток, который нарушает клеточный реполяризация.[21]
LQT10 - чрезвычайно редкий подтип, вызванный вариантами в SCN4B ген. Продуктом этого гена является вспомогательная бета-субъединица (NaVβ4), образующие сердечные натриевые каналы, варианты которых увеличивают поздний устойчивый натриевый ток.[21] LQT13 вызван вариантами в ГИРК4, белок, участвующий в парасимпатической модуляции сердца.[21] Клинически пациенты характеризуются лишь умеренным удлинением интервала QT, но повышенной склонностью к предсердным аритмиям. LQT14, LQT15 и LQT16 вызываются вариантами генов, ответственных за кальмодулин (CALM1, CALM2, и CALM3 соответственно).[21] Кальмодулин взаимодействует с несколькими ионными каналами, и его роли включают модуляцию кальциевого тока L-типа в ответ на концентрации кальция и транспортировку белков, продуцируемых KCNQ1 и тем самым влияя на калиевые токи.[21] Точные механизмы, с помощью которых эти генетические варианты удлиняют интервал QT, остаются неясными.[21]
Синдром Джервелла и Ланге – Нильсена
Синдром Джервелла и Ланге – Нильсена (JLNS) - редкая форма LQTS, наследуемая по аутосомно-рецессивному типу. Помимо значительного удлинения интервала QT, пациенты рождаются с тяжелой нейросенсорной глухотой, поражающей оба уха. Синдром вызван наследованием двух копий определенного варианта в KCNE1 или KCNQ1 гены. Одни и те же генетические варианты приводят к формам LQT1 и LQT5 синдрома Романо-Уорда, если наследуется только одна копия варианта.[10] JLNS обычно ассоциируется с более высоким риском аритмий, чем большинство других форм LQTS.[4]
Синдром Андерсена – Тавила (LQT7)
LQT7, также известный как Синдром Андерсена-Тавила, характеризуется триадой признаков - помимо удлиненного интервала QT, пораженные могут испытывать перемежающуюся слабость, часто возникающую при низких концентрациях калия в крови (периодический гипокалиемический паралич), а также характерные лицевые и скелетные аномалии, такие как небольшая нижняя челюсть (микрогнатия ), низко посаженные уши и сросшиеся или неправильно расположенные пальцы рук и ног (синдактилия и клинодактилия ). Это заболевание наследуется по аутосомно-доминантному типу и часто связано с мутациями в KCNJ2 ген, который кодирует белок K калиевого каналаir2.1.[23]
Синдром Тимоти (LQT8)
LQT8, также известный как Синдром Тимоти сочетает удлиненный интервал QT со сращением пальцев рук или ног (синдактилия). Часто наблюдаются аномалии структуры сердца, в том числе: дефект межжелудочковой перегородки, тетралогия Фалло, и гипертрофическая кардиомиопатия.[24][25] Состояние проявляется в раннем возрасте, и средняя продолжительность жизни составляет 2,5 года, причем смерть чаще всего вызывается желудочковой аритмией. Многие дети с синдромом Тимоти, которые выживают дольше этого срока, имеют особенности расстройство аутистического спектра. Синдром Тимоти вызывается вариантами кальциевого канала Cav1.2, кодируемого геном CACNA1c.[26]
Таблица связанных генов
Ниже приводится список генов, связанных с LQTS:
| Тип | OMIM | Ген | Заметки |
| LQT1 | 192500 | KCNQ1 | Кодирует α-субъединицу калиевого канала медленного выпрямителя с задержкой KV7.1 проводящий калиевый ток яKs.[19] |
| LQT2 | 152427 | KCNH2 | Также известен как hERG. Кодирует α-субъединицу калиевого канала быстродействующего выпрямителя KV11.1 проводящий калиевый ток яKr.[19] |
| LQT3 | 603830 | SCN5A | Кодирует α-субъединицу сердечного натриевого канала NaV1.5 проводящий натриевый ток яNa.[19] |
| LQT4 | 600919 | ANK2 | Кодирует анкирин B, который закрепляет ионные каналы в клетке. Спорный вопрос о том, действительно ли ген чувствительности вызывает заболевание, или ген малой чувствительности QT.[19] |
| LQT5 | 176261 | KCNE1 | Кодирует MinK, β-субъединицу калиевого канала. Гетерозиготное наследование вызывает синдром Романо – Уорда, гомозиготное наследование вызывает синдром Джервелла и Ланге – Нильсена.[19] |
| LQT6 | 603796 | KCNE2 | Кодирует MiRP1, β-субъединицу калиевого канала. Спорный вопрос о том, действительно ли ген чувствительности вызывает заболевание, или ген малой чувствительности QT.[19] |
| LQT7 | 170390 | KCNJ2 | Кодирует входящий выпрямительный калиевый ток Kir2.1 проводящий калиевый ток яK1. Причины Синдром Андерсена-Тавила.[19] |
| LQT8 | 601005 | CACNA1c | Кодирует α-субъединицу CaV1.2 из кальциевый канал Cav1.2, несущий кальциевый ток яCa (L). Причины Синдром Тимоти.[19] |
| LQT9 | 611818 | CAV3 | Кодирует кавеолин-3, отвечающий за образование мембранных мешочков, известных как кавеолы. Мутации в этом гене могут увеличить поздний ток натрия. яNa.[19] |
| LQT10 | 611819 | SCN4B | Кодирует β4-субъединицу сердечного натриевого канала.[19] |
| LQT11 | 611820 | AKAP9 | Кодирует белок, связанный с A-киназой, который взаимодействует с KV7.1.[19] |
| LQT12 | 601017 | SNTA1 | Кодирует синтрофин-α1. Мутации в этом гене могут увеличить поздний ток натрия. яNa.[19] |
| LQT13 | 600734 | KCNJ5 | Также известен как GIRK4, кодирует чувствительные к белку G, выпрямляющие внутрь калиевые каналы (Kir3.4), по которым протекает калиевый ток яК (АЧ).[19] |
| LQT14 | 616247 | CALM1 | Кодирует кальмодулин-1, кальций-связывающий белок-мессенджер, который взаимодействует с кальциевым током. яCa (L).[19] |
| LQT15 | 616249 | CALM2 | Кодирует кальмодулин-2, кальций-связывающий белок-мессенджер, который взаимодействует с кальциевым током. яCa (L).[19] |
| LQT16 | 114183 | CALM3 | Кодирует кальмодулин-3, кальций-связывающий белок-мессенджер, который взаимодействует с кальциевым током. яCa (L).[19] |
Приобретено
Хотя синдром удлиненного интервала QT часто является генетическим заболеванием, удлиненный интервал QT, связанный с повышенным риском нарушения сердечного ритма, также может возникать у людей без генетических отклонений, обычно из-за побочного действия лекарств. Удлинение интервала QT, вызванное лекарствами часто является результатом лечения антиаритмический наркотики, такие как амиодарон и соталол, антибиотики, такие как эритромицин, или антигистаминные препараты такие как терфенадин.[17] К другим препаратам, удлиняющим интервал QT, относятся: нейролептики такие как галоперидол и зипразидон, а антидепрессант циталопрам.[27][16] Списки лекарств, связанных с удлинением интервала QT, таких как CredibleMeds базу данных можно найти в Интернете.[28]
Другие причины приобретенного LQTS включают аномально низкий уровень калия (гипокалиемия ) или магний (гипомагниемия ) в крови. Это может усугубиться после внезапного уменьшения кровоснабжения сердца (инфаркт миокарда ), низкий уровень гормона щитовидной железы (гипотиреоз ) и медленное сердцебиение (брадикардия ).[29]
Нервная анорексия был связан с внезапной смертью, возможно, из-за удлинения интервала QT. Недоедание, наблюдаемое при этом состоянии, иногда может влиять на концентрацию в крови солей, таких как калий, что потенциально может привести к приобретенному синдрому удлиненного интервала QT, что в свою очередь вызывает внезапная сердечная смерть. Недоедание и связанные с ним изменения солевого баланса развиваются в течение длительного периода времени, и быстрое возобновление питания может еще больше нарушить солевой дисбаланс, увеличивая риск аритмий. Поэтому необходимо следить за уровнем электролитов, чтобы избежать осложнений. синдром возобновления питания.[30]
Факторы, удлиняющие интервал QT, являются аддитивными, что означает, что комбинация факторов (таких как прием препаратов, удлиняющих интервал QT и низкий уровень калия), может вызывать большее удлинение интервала QT, чем каждый фактор по отдельности. Это также относится к некоторым генетическим вариантам, которые сами по себе лишь минимально удлиняют интервал QT, но могут сделать людей более восприимчивыми к значительному удлинению интервала QT, вызванному лекарствами. [29]
Механизмы
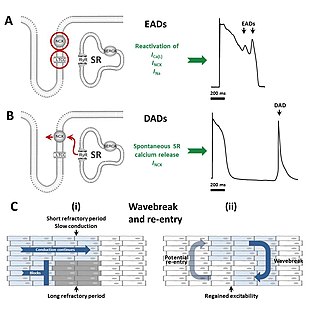
Различные формы синдрома удлиненного интервала QT, как врожденные, так и приобретенные, вызывают аномальные сердечные ритмы (аритмии), влияя на электрические сигналы, используемые для координации отдельных клеток сердца. Общая тема - продолжение потенциал сердечного действия - характерный образец изменения напряжения на клеточной мембране, возникающий при каждом ударе сердца.[10] Клетки сердца в расслабленном состоянии обычно имеют меньше положительно заряженных ионы на внутренней стороне их клеточная мембрана чем на внешней стороне, называемой мембраной поляризованный. Когда клетки сердца договор положительно заряженные ионы, такие как натрий и кальций, входят в клетку, выравнивая или меняя полярность, или деполяризующий сотовый. После того, как произошло сокращение, клетка восстанавливает свою полярность (или переполяризует), позволяя положительно заряженным ионам, таким как калий, покидать клетку, возвращая мембрану в расслабленное, поляризованное состояние. При синдроме удлиненного QT реполяризации требуется больше времени, что проявляется в отдельных клетках как более длительный потенциал действия, а на поверхности ЭКГ отмечается как длинный интервал QT.[10]
Продленные потенциалы действия могут приводить к аритмиям по нескольким причинам. Аритмия, характерная для синдрома удлиненного интервала QT, Торсад де Пуант, начинается, когда первоначальный потенциал действия вызывает дальнейшие аномальные потенциалы действия в виде постдеполяризации. Ранняя постдеполяризация, происходящая до того, как клетка полностью реполяризовалась, особенно часто наблюдается, когда потенциалы действия продлены, и возникают из-за реактивации кальция и натрия. каналы обычно он отключается до следующего пульса.[31] При правильных условиях реактивация этих токов, чему способствует обменник натрия-кальция, может вызвать дальнейшую деполяризацию клетки.[31] Ранние постдеполяризации, вызывающие аритмию при синдроме удлиненного интервала QT, как правило, возникают из-за Волокна Пуркинье сердечной проводящей системы.[32] Ранняя постдеполяризация может происходить как единичные события, но может происходить неоднократно, что приводит к множественной быстрой активации клетки.[31]
Некоторые исследования показывают, что отсроченная постдеполяризация, возникающая после завершения реполяризации, также может играть роль в синдроме удлиненного интервала QT.[32] Эта форма постдеполяризации возникает из-за спонтанного высвобождения кальция из внутриклеточного хранилища кальция, известного как саркоплазматический ретикулум, вытесняя кальций из клетки через обменник кальций натрия в обмен на натрий, генерирующий чистый внутренний ток.[31]
Хотя есть убедительные доказательства того, что триггером Torsades de Pointes является постдеполяризация, менее ясно, что поддерживает эту аритмию. Некоторые данные свидетельствуют о том, что повторяющиеся постдеполяризации из многих источников способствуют продолжающейся аритмии.[32] Однако некоторые предполагают, что аритмия поддерживается за счет механизма, известного как повторный вход. Согласно этой модели, пролонгация потенциала действия происходит в разной степени в разных слоях организма. сердечная мышца с более длинными потенциалами действия в одних слоях, чем в других.[32] В ответ на запускающий импульс волны деполяризации будут распространяться по областям с более короткими потенциалами действия, но блокироваться в областях с более длинными потенциалами действия. Это позволяет деполяризующему волновому фронту огибать участки блока, потенциально образуя замкнутую петлю и самовоспроизводясь. Характер скручивания на ЭКГ можно объяснить движением ядра возвратного контура в виде меандрирующего спиральная волна.[32]
Диагностика


Диагностировать синдром удлиненного интервала QT непросто. В то время как отличительной чертой LQTS является удлинение интервала QT, интервал QT сильно варьируется как у здоровых, так и у тех, у кого есть LQTS. Это приводит к перекрытию интервалов QT у тех, у кого есть LQTS и нет. У 2,5% людей с генетически подтвержденным LQTS интервал QT находится в пределах нормы.[20] И наоборот, учитывая нормальное распределение интервалов QT, часть здоровых людей будет иметь более длинный интервал QT, чем любое произвольное ограничение.[20] Поэтому при постановке диагноза следует принимать во внимание другие факторы, выходящие за пределы интервала QT, некоторые из которых были включены в системы оценки.[4]
ЭКГ

Синдром удлиненного интервала QT в основном диагностируется путем измерения QT интервал с поправкой на частоту сердечных сокращений (QTc) на электрокардиограмме (ЭКГ) в 12 отведениях. Синдром удлиненного интервала QT связан с удлинением интервала QTc, хотя в некоторых генетически подтвержденных случаях LQTS это удлинение может быть скрытым, известным как скрытый LQTS.[20] QTc составляет менее 450 мс у 95% нормальных мужчин и менее 460 мсек у 95% нормальных женщин. LQTS предлагается, если QTc длиннее этих пороговых значений. Однако, поскольку 5% нормальных людей также попадают в эту категорию, некоторые предлагают пороговые значения 470 и 480 мс для мужчин и женщин соответственно, что соответствует 99-й центили нормальных значений.[20]
Основные подтипы унаследованного LQTS связаны с конкретными особенностями ЭКГ. LQT1 обычно ассоциируется с широким спектром Зубцы Т, в то время как зубцы T в LQT2 зазубрены и имеют меньшую амплитуду, в то время как в LQT3 зубцы T часто появляются поздно, им предшествует длинный изоэлектрический сегмент.[20]
Оценка Шварца

Шкала Шварца была предложена как метод объединения клинических факторов и факторов ЭКГ для оценки вероятности наличия у человека наследственной формы LQTS.[7] В таблице ниже перечислены критерии, используемые для расчета баллов.
| Шкала Шварца для диагностики наследственного синдрома удлиненного интервала QT. [33] | |||
| Скорректированный интервал QT (QTc) | ≥ 480 мс | 3 балла | QTc определяется согласно Поправка Базетт |
| 460–470 мс | 2 балла | ||
| 450 мс и мужской пол | 1 балл | ||
| Торсады-де-пуанты | 2 балла | ||
| Альтернативные зубцы Т | 1 балл | ||
| Зубцы T с зубцами как минимум в 3 отведениях | 1 балл | ||
| Низкая частота пульса для возраста (дети) | 0,5 балла | ||
| Обморок | со стрессом | 2 балла | Не может получить баллы как за обморок, так и за торсады |
| без стресса | 1 балл | ||
| Врожденная глухота | 0,5 балла | ||
| История семьи | Другой член семьи с подтвержденным LQTS | 1 балл | Один и тот же член семьи не может быть засчитан для LQTS и внезапной смерти |
| Внезапная сердечная смерть у ближайшего родственника в возрасте <30 лет | 0,5 балла | ||
| Оценка: 0–1: низкая вероятность LQTS; 2–3: промежуточная вероятность LQTS; ≥ 4: высокая вероятность LQTS | |||
Прочие расследования
В случаях диагностической неопределенности могут быть полезны другие исследования, направленные на выявление удлиненного интервала QT. В дополнение к своему влиянию на интервал QT в покое LQTS влияет на то, как изменяется QT в ответ на упражнения и стимуляцию катехоламинами, такими как адреналин. Для выявления этих аномальных реакций можно использовать провокационные тесты в виде тестов на толерантность к физической нагрузке или прямого введения адреналина.[34] Эти исследования наиболее полезны для определения группы лиц с врожденным LQTS типа 1 (LQT1), у которых в покое нормальный интервал QT. У этих людей упражнения или инфузия адреналина могут привести к парадоксальному удлинению интервала QT, раскрывая основное состояние.[20]
Рекомендации по сокращению
Международные согласованные руководящие принципы различаются по степени удлинения интервала QT, необходимого для диагностики LQTS. В Европейское общество кардиологов рекомендует, с симптомами или без них или другими исследованиями, LQTS можно диагностировать, если скорректированный интервал QT превышает 480 мс. Они рекомендуют рассматривать диагноз при наличии QTc более 460 мс, если произошли необъяснимые обмороки.[4] В Общество сердечного ритма руководящие принципы являются более строгими и рекомендуют отсечку QTc более 500 мс при отсутствии других факторов, удлиняющих QT, или более 480 мс при обмороке.[5] Оба набора руководств согласны с тем, что LQTS также может быть диагностирован, если у человека оценка по Шварцу выше 3 или если идентифицирован патогенный генетический вариант, связанный с LQTS, независимо от интервала QT.[4][5]
лечение
Тем, у кого диагностирован LQTS, обычно рекомендуется избегать лекарств, которые могут еще больше продлить интервал QT или снизить порог для TDP, списки которых можно найти в общедоступные онлайн-базы данных.[35] В дополнение к этому, для людей с LQTS известны два варианта вмешательства: предотвращение аритмии и прекращение аритмии.
Профилактика аритмии
Подавление аритмии включает использование лекарств или хирургических процедур, которые воздействуют на основную причину аритмий, связанных с LQTS. Поскольку причиной аритмий в LQTS является ранняя постдеполяризация (EAD), и они усиливаются в состояниях адренергической стимуляции, могут быть предприняты шаги для подавления адренергической стимуляции у этих людей. К ним относятся администрирование агенты, блокирующие бета-рецепторы, что снижает риск возникновения аритмий, вызванных стрессом. Бета-блокаторы являются эффективным средством лечения LQTS, вызванного LQT1 и LQT2.[7]
Генотип и продолжительность интервала QT являются независимыми предикторами рецидива угрожающих жизни событий во время терапии бета-адреноблокаторами. Чтобы быть конкретным, наличие QTc> 500 мс и генотипов LQT2 и LQT3 связано с самой высокой частотой рецидивов. У этих пациентов первичная профилактика с использованием имплантируемые кардиовертеры-дефибрилляторы можно считать.[7]
- Добавки калия: если содержание калия в крови повышается, потенциал действия сокращается, поэтому повышение концентрации калия может свести к минимуму возникновение аритмий. Он должен работать лучше всего в LQT2, поскольку канал hERG особенно чувствителен к концентрации калия, но его использование является экспериментальным, а не доказательным.
- Препараты, блокирующие натриевые каналы, такие как мексилетин использовались для предотвращения аритмий при синдроме удлиненного интервала QT.[36] Хотя наиболее убедительным показанием является тот, у кого синдром удлиненного QT вызван дефектными натриевыми каналами, производящими устойчивый поздний ток (LQT3), мексилетин также укорачивает интервал QT при других формах синдрома удлиненного QT, включая LQT1, LQT2 и LQT8.[36] Поскольку преобладающее действие мексилетина связано с ранним пиковым натриевым током, существуют теоретические причины, по которым препараты, которые преимущественно подавляют поздний натриевый ток, такие как ранолазин может быть более эффективным, хотя свидетельств того, что это так на практике, мало.[36]
- Ампутация шейная симпатическая цепь (осталось стеллэктомия ). Эта терапия обычно предназначена для LQTS, вызванного JLNS,[7] но в некоторых случаях может использоваться в качестве дополнительной терапии к бета-адреноблокаторам. В большинстве случаев современная терапия отдает предпочтение имплантации ИКД, если терапия бета-блокаторами не дает результатов.
Прекращение аритмии
Прекращение аритмии предполагает прекращение опасной для жизни аритмии, если она уже произошла. Одной из эффективных форм купирования аритмии у людей с LQTS является установка имплантируемого кардиовертера-дефибриллятора (ICD). Также для восстановления синусового ритма можно использовать внешнюю дефибрилляцию. ИКД обычно используются у пациентов с эпизодами обморока, несмотря на терапию бета-блокаторами, а также у пациентов, перенесших остановку сердца.
Надеемся, что благодаря лучшему знанию генетики, лежащей в основе LQTS, станут доступны более точные методы лечения.[37]
Результаты
Для людей, которые испытывают остановку сердца или обмороки, вызванные LQTS, и которые не получают лечения, риск смерти в течение 15 лет составляет около 50%.[8] При тщательном лечении этот показатель снижается до менее 1% за 20 лет.[3] Те, у кого проявляются симптомы до 18 лет, с большей вероятностью испытают остановку сердца.[20][38]
Эпидемиология
Унаследованный LQTS, по оценкам, затрагивает от одного из 2500 до 7000 человек.[7]
История
Первый задокументированный случай LQTS был описан в Лейпциг Мейснера в 1856 году, когда глухая девушка умерла после того, как на нее накричал учитель. Вскоре после уведомления родители девочки сообщили, что ее старший брат, также глухой, ранее скончался после ужасного испуга.[39] Это было за несколько десятилетий до изобретения ЭКГ, но, вероятно, это первый описанный случай синдрома Джервелла и Ланге-Нильсена. В 1957 году первый случай, задокументированный с помощью ЭКГ, описал Антон Джервелл и Фред Ланге-Нильсен, работает в Tønsberg, Норвегия.[40] Итальянский педиатр Чезарино Романо, в 1963 году,[41] и ирландский педиатр Оуэн Конор Уорд в 1964 г.[42] отдельно описан более распространенный вариант LQTS с нормальным слухом, позже названный синдромом Романо-Уорда. Создание Международного реестра синдромов удлиненного интервала QT в 1979 г. позволило множеству родословные подлежат всесторонней оценке. Это помогло обнаружить многие из множества задействованных генов.[43]
использованная литература
- ^ а б c d е ж «Синдром удлиненного интервала QT». Информационный центр по генетическим и редким заболеваниям (GARD) - программа NCATS. 2017. Получено 14 декабря 2017.
- ^ а б c Морита Х., Ву Дж., Зипес Д.П. (август 2008 г.). «Синдромы QT: длинный и короткий». Ланцет. 372 (9640): 750–63. Дои:10.1016 / S0140-6736 (08) 61307-0. PMID 18761222. S2CID 41181673.
- ^ а б c d е Ферри Ф.Ф. (2016). Электронная книга Ferri's Clinical Advisor 2017: 5 книг в 1. Elsevier Health Sciences. п. 736. ISBN 9780323448383.
- ^ а б c d е ж г час Приори С.Г., Бломстрём-Лундквист С., Маццанти А., Блом Н., Борггрефе М., Камм Дж. И др. (Ноябрь 2015 г.). «Рекомендации ESC 2015 по ведению пациентов с желудочковой аритмией и профилактике внезапной сердечной смерти: Целевая группа по ведению пациентов с желудочковой аритмией и профилактике внезапной сердечной смерти Европейского общества кардиологов (ESC) При поддержке: Ассоциация европейской детской и врожденной кардиологии (AEPC) ». Europace. 17 (11): 1601–87. Дои:10.1093 / europace / euv319. PMID 26318695.
- ^ а б c d Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C и др. (Октябрь 2013). «Краткое изложение: консенсусное заявление экспертов HRS / EHRA / APHRS по диагностике и ведению пациентов с наследственными синдромами первичной аритмии». Europace. 15 (10): 1389–406. Дои:10.1093 / europace / eut272. PMID 23994779.
- ^ а б c d е ж г час я j k «Синдром удлиненного интервала QT». NHLBI, NIH. Получено 14 декабря 2017.
- ^ а б c d е ж г Левин Э., Росеро С.З., Будзиковски А.С., Мосс Эй Джей, Zareba W, Daubert JP (август 2008 г.). «Врожденный синдром удлиненного интервала QT: соображения для врачей первичного звена». Кливлендский медицинский журнал клиники. 75 (8): 591–600. Дои:10.3949 / ccjm.75.8.591. PMID 18756841. S2CID 4237579.
- ^ а б Акерман М.Дж., Приори С.Г., Дубин А.М., Коуи П., Линкер Н.Дж., Слотвинер Д. и др. (Январь 2017 г.). «Бета-адреноблокаторы при синдроме удлиненного интервала QT и катехоламинергической полиморфной желудочковой тахикардии: все ли бета-адреноблокаторы эквивалентны?». Сердечного ритма. 14 (1): e41 – e44. Дои:10.1016 / j.hrthm.2016.09.012. PMID 27659101.
Среди пациентов, у которых возникло сердечное событие, вызванное LQTS (аритмический обморок, аритмический обморок с последующими судорогами или прерывание сердечного приступа), нелеченая естественная история мрачна, с> 50% летальностью через 15 лет.

- ^ Винсент Дж, Абрахам Э., Кочанек П., Мур Ф.А., Финк МП (2011). Электронная книга "Учебник по интенсивной терапии". Elsevier Health Sciences. п. 578. ISBN 978-1437715682.
- ^ а б c d е ж г час Tester DJ, Schwartz PJ, Ackerman MJ (2013). «Врожденный синдром удлиненного интервала QT». В Gussak I, Antzelevitch C (ред.). Электрические болезни сердца. Лондон: Спрингер. С. 439–468. Дои:10.1007/978-1-4471-4881-4_27. ISBN 978-1-4471-4881-4.
- ^ Макмиллан Дж. А., Фейгин Р. Д., ДеАнгелис С., Джонс М. Д. (2006). Педиатрия Оски: принципы и практика. Липпинкотт Уильямс и Уилкинс. п. 1677. ISBN 978-0-7817-3894-1.
- ^ а б Мадан Н., Карвалью К.С. (февраль 2017 г.). «Неврологические осложнения сердечных заболеваний». Семинары по детской неврологии. 24 (1): 3–13. Дои:10.1016 / j.spen.2017.01.001. PMID 28779863.
Обморок может привести к судорогам и его легко спутать с эпилептическими припадками.
- ^ Накадзима Т., Канеко Ю., Курабаяши М. (2015). «Выявление специфических триггеров и провоцирующих факторов фатальных сердечных событий при синдромах наследственной аритмии». Тираж Журнал. 79 (6): 1185–92. Дои:10.1253 / circj.CJ-15-0322. PMID 25925977.
- ^ а б c Тринкли К.Е., Пейдж Р.Л., Лиен Х., Ямануе К., Тисдейл Дж. Э. (декабрь 2013 г.). «Удлинение интервала QT и риск torsades de pointes: главное для клиницистов». Текущие медицинские исследования и мнения. 29 (12): 1719–26. Дои:10.1185/03007995.2013.840568. PMID 24020938. S2CID 206967580.
- ^ а б Баршешет А., Доценко О., Гольденберг И. (ноябрь 2013 г.). «Стратификация риска по генотипу и ведение пациентов с синдромом удлиненного интервала QT». Анналы неинвазивной электрокардиологии. 18 (6): 499–509. Дои:10.1111 / anec.12117. ЧВК 6932574. PMID 24206565.
- ^ а б c d Роден Д.М. (март 2004 г.). «Медикаментозное удлинение интервала QT». Медицинский журнал Новой Англии. 350 (10): 1013–22. Дои:10.1056 / NEJMra032426. PMID 14999113. S2CID 15251057.
- ^ а б Томсон С., Райт П. (2014-10-15). «Синдром удлиненного интервала QT». Фармацевтический журнал. 293 (7833). Получено 18 октября 2014.
- ^ Хедли П.Л., Йоргенсен П., Шламовиц С., Вангари Р., Мулман-Смук Дж., Бринк П.А. и др. (Ноябрь 2009 г.). «Генетическая основа синдромов длинного QT и короткого QT: обновление мутации». Человеческая мутация. 30 (11): 1486–511. Дои:10.1002 / humu.21106. PMID 19862833. S2CID 19122696.
- ^ а б c d е ж г час я j k л м п о п q р s Джудичесси-младший, Уайлд А.А., Акерман М.Дж. (октябрь 2018 г.). «Генетическая архитектура синдрома удлиненного интервала QT: критическая переоценка». Тенденции в сердечно-сосудистой медицине. 28 (7): 453–464. Дои:10.1016 / j.tcm.2018.03.003. ЧВК 6590899. PMID 29661707.
- ^ а б c d е ж г час я Джудичесси-младший, Акерман М.Дж. (октябрь 2013 г.). «Управление врожденным синдромом удлиненного интервала QT на основе генотипа и фенотипа». Актуальные проблемы кардиологии. 38 (10): 417–55. Дои:10.1016 / j.cpcardiol.2013.08.001. ЧВК 3940076. PMID 24093767.
- ^ а б c d е ж г час я j k л м п о п q Bohnen MS, Peng G, Robey SH, Terrenoire C, Iyer V, Sampson KJ, Kass RS (январь 2017 г.). «Молекулярная патофизиология врожденного синдрома удлиненного интервала QT». Физиологические обзоры. 97 (1): 89–134. Дои:10.1152 / Physrev.00008.2016. ЧВК 5539372. PMID 27807201.
- ^ Бьеррегаард П. (август 2018 г.). «Диагностика и лечение синдрома короткого интервала QT». Сердечного ритма. 15 (8): 1261–1267. Дои:10.1016 / j.hrthm.2018.02.034. PMID 29501667.
- ^ Нгуен Х.Л., Пипер Г.Х., Вилдерс Р. (декабрь 2013 г.). «Синдром Андерсена-Тавиля: клинические и молекулярные аспекты». Международный журнал кардиологии. 170 (1): 1–16. Дои:10.1016 / j.ijcard.2013.10.010. PMID 24383070.
- ^ Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Stephens K и др. (Февраль 2006 г.). «Синдром Тимоти». GeneReviews. Сиэтл (Вашингтон): Вашингтонский университет, Сиэтл. PMID 20301577.
- ^ «Синдром Тимоти». PMID 20301577. Цитировать журнал требует
| журнал =(Помогите) - ^ Тристани-Фирузи М, Этеридж СП (2013). Гусак I, Анцелевич С (ред.). Синдромы Андерсена-Тавила и Тимоти. Электрические болезни сердца. Springer London. С. 561–567. Дои:10.1007/978-1-4471-4881-4_32. ISBN 978-1-4471-4880-7.
- ^ Beach SR, Celano CM, Noseworthy PA, Januzzi JL, Huffman JC (январь 2013 г.). «Удлинение интервала QT, пуанты и психотропные препараты». Психосоматика. 54 (1): 1–13. Дои:10.1016 / j.psym.2012.11.001. PMID 23295003.
- ^ Woosley RL, Black K, Heise CW, Romero K (февраль 2018 г.). "CredibleMeds.org: Что он предлагает?" (PDF). Тенденции в сердечно-сосудистой медицине. 28 (2): 94–99. Дои:10.1016 / j.tcm.2017.07.010. HDL:10150/627826. PMID 28801207.
- ^ а б Эль-Шериф N, Turitto G, Boutjdir M (апрель 2018 г.). «Приобретенный синдром удлиненного интервала QT и torsade de pointes». Электрокардиостимуляция и клиническая электрофизиология. 41 (4): 414–421. Дои:10.1111 / pace.13296. PMID 29405316. S2CID 46795997.
- ^ Хауреги-Гарридо Б., Хауреги-Лобера I (февраль 2012 г.). «Внезапная смерть при расстройствах пищевого поведения». Здоровье сосудов и управление рисками. 8: 91–8. Дои:10.2147 / VHRM.S28652. ЧВК 3292410. PMID 22393299.
- ^ а б c d Wit AL (июнь 2018 г.). «Постдеполяризации и триггерная активность как механизм клинических аритмий». Электрокардиостимуляция и клиническая электрофизиология. 41 (8): 883–896. Дои:10.1111 / pace.13419. PMID 29920724. S2CID 49310809.
- ^ а б c d е Эль-Шериф N, Turitto G, Boutjdir M (май 2019 г.). «Приобретенный синдром удлиненного интервала QT и электрофизиология Torsade de Pointes». Обзор аритмии и электрофизиологии. 8 (2): 122–130. Дои:10.15420 / aer.2019.8.3. ЧВК 6528034. PMID 31114687.
- ^ Шварц П.Дж., Мосс А.Дж., Винсент Г.М., Крэмптон Р.С. (август 1993 г.). «Диагностические критерии синдрома удлиненного интервала QT. Обновление». Тираж. 88 (2): 782–4. Дои:10.1161 / 01.CIR.88.2.782. PMID 8339437.
- ^ Обейесекере М.Н., Кляйн Г.Дж., Моди С., Леонг-Сит П., Гула Л.Дж., Йи Р. и др. (Декабрь 2011 г.). «Как проводить и интерпретировать провокационные тесты для диагностики синдрома Бругада, синдрома удлиненного интервала QT и катехоламинергической полиморфной желудочковой тахикардии». Тираж. Аритмия и электрофизиология. 4 (6): 958–64. Дои:10.1161 / CIRCEP.111.965947. PMID 22203660.
- ^ «Список препаратов QT по группам риска». Аризонский центр образования и исследований в области терапии. Архивировано из оригинал 24 декабря 2010 г.. Получено 2010-07-04.
- ^ а б c Ли Г, Чжан Л. (ноябрь 2018 г.). «Роль мексилетина в лечении синдрома удлиненного интервала QT». Журнал электрокардиологии. 51 (6): 1061–1065. Дои:10.1016 / j.jelectrocard.2018.08.035. PMID 30497731.
- ^ Комптон С.Дж., Люкс Р.Л., Рэмси М.Р., Стрелих К.Р., Сангинетти М.С., Грин Л.С. и др. (Сентябрь 1996 г.). «Генетически определенная терапия наследственного синдрома удлиненного интервала QT. Коррекция аномальной реполяризации калием». Тираж. 94 (5): 1018–22. Дои:10.1161 / 01.CIR.94.5.1018. PMID 8790040.
- ^ «Связь генотипа и риска».
- ^ Tranebjaerg L, Bathen J, Tyson J, Bitner-Glindzicz M (сентябрь 1999 г.). «Синдром Джервелла и Ланге-Нильсена: норвежская перспектива». Американский журнал медицинской генетики. 89 (3): 137–46. Дои:10.1002 / (SICI) 1096-8628 (19990924) 89: 3 <137 :: AID-AJMG4> 3.0.CO; 2-C. PMID 10704188.
- ^ Джервелл А., Ланге-Нильсен Ф (июль 1957 г.). «Врожденный глухонемой, функциональный порок сердца с удлинением интервала Q-T и внезапная смерть». Американский журнал сердца. 54 (1): 59–68. Дои:10.1016/0002-8703(57)90079-0. PMID 13435203.
- ^ Romano C, Gemme G, Pongiglione R (сентябрь 1963 г.). «[Арритмии детского возраста. II. Синкопальные приступы из-за пароксизмальной фибрилляции желудочков. (Презентация первого случая в итальянской педиатрической литературе]». La Clinica Pediatrica (на итальянском). 45: 656–83. PMID 14158288.
- ^ Ward OC (апрель 1964 г.). «Новый семейный сердечный синдром у детей». Журнал Ирландской медицинской ассоциации. 54: 103–6. PMID 14136838.
- ^ Мосс А.Дж., Шварц П.Дж. (март 2005 г.). «25-летие Международного реестра синдромов длинного интервала QT: постоянные поиски секретов синдрома длинного интервала QT». Тираж. 111 (9): 1199–201. Дои:10.1161 / 01.CIR.0000157069.91834.DA. PMID 15753228.
{kind=link}
- Заметки
- Гольдман Л (2011). Goldman's Cecil Medicine (24-е изд.). Филадельфия: Эльзевьер Сондерс. п. 1196. ISBN 978-1437727883.
внешние ссылки
| Классификация | |
|---|---|
| Внешние ресурсы |
- CredibleMeds.org, содержит список препаратов, удлиняющих интервал QT